@addx
I'm not sure if a single superordinated aging promotor really exists, but If so, the hypothalamus would be the first candidate.
Im not saying that there's just one.
I'm saying kappa and mu opioid mechanisms modulate responsive/adaptive changes to neuronal growth/differentiation rate before maturity, they trigger adaptive changes to synaptic schemas during lifetime, they trigger adaptive muscle changes during lifetime, trigger skin tissue adaptive changes and even sperm adaptive changes all in response to experience, maybe more..
Maturing is guided by a complex cascade of cells diffentiating, producing proteins which cause futher and different differentiation, tissues producing "counter tissues". An insect can duplicate only gene in the HOX region and it might cause him a fully functional set of wings or legs, meaning one gene duplication in the base "segment" region of DNA will have the muscle and nervous system genes fully work on the extra set, connect it to the brain and the brain even adjust to using them. So, the process of producing the body as a connected set of tissues is very "fractalish" and complex as can be imagined from that example.
Adaptive changes in response to "active life experience" however seem to be neatly modulated mostly by opioidergic effects. Opioidergic effects are affected by a different number of cofactors and depending on which cell culture.
Kappa opioid activation has different effects depending p38 mapk activation which is a major stress pathway (as proposed fear-analgesia - effects on synaptic schemas) - https://hopecenter.wustl.edu/?p=3460
http://www.ncbi.nlm....pubmed/10807318
MAP kinase pathways activated by stress: the p38 MAPK pathway.
Abstract
A stress-activated serine/threonine protein kinase, p38 mitogen-activated protein kinase (p38 MAPK), belongs to the MAP kinase superfamily. Diverse extracellular stimuli, including ultraviolet light, irradiation, heat shock, high osmotic stress, proinflammatory cytokines and certain mitogens, trigger a stress-regulated protein kinase cascade culminating in activation of p38 MAPK through phosphorylation on a TGY motif within the kinase activation loop. p38 MAPK appears to play a major role in apoptosis, cytokine production, transcriptional regulation, and cytoskeletal reorganization, and has been causally implicated in sepsis, ischemic heart disease, arthritis, human immunodeficiency virus infection, and Alzheimer's disease. The availability of specific inhibitors helps to clarify the role that p38 MAPK plays in these processes, and may ultimately offer therapeutic benefit for certain critically ill patients
Arrestin-mediated activation of
p38 MAPK: molecular mechanisms and behavioral consequences.
Abstract
Studies of kappa opioid receptor signaling mechanisms during the last decade have demonstrated that agonist activation of the receptor results in Gβγ-dependent signaling and distinct arrestin-dependent signaling events. Gβγ-dependent signaling results in ion channel regulation causing neuronal inhibition, inhibition of transmitter release, and subsequent analgesic responses. In contrast, arrestin-dependent signaling events result inp38 MAPK activation and subsequent dysphoric and proaddictive behavioral responses. Resolution of these two branches of signaling cascades has enabled strategies designed to identify pathway-selective drugs that may have unique therapeutic utilities.
http://www.ncbi.nlm....pubmed/24292835
Possible role of dynorphins in Alzheimer's disease and age-related cognitive deficits.
Abstract
BACKGROUND/AIMS:
Expression of dynorphin, an endogenous opioid peptide, increases with age and has been associated with cognitive deficits in rodents. Elevated dynorphin levels have been reported in postmortem samples from Alzheimer's disease (AD) patients, and prodynorphin (PDYN) gene polymorphisms might be linked to cognitive function in the elderly. Activation of κ-opioid receptors by dynorphins has been associated with stress-related memory impairments. Interestingly, these peptides can also modulate glutamate neurotransmission and may affect synaptic plasticity underlying memory formation. N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazol-propionate (AMPA) ionotropic glutamate receptor levels generally decrease with aging, and their function is impaired in AD.
METHODS:
Here, we compared the impact of aging on ionotropic glutamate receptor levels in the hippocampal formation of wild-type (WT) and Pdyn knock-out (KO) mice.
RESULTS:
We observed a significant reduction in GluR1 and GluR2 AMPA receptor subunits in the hippocampal formation of 18- to 25-month-old WT mice in comparison with 6-month-old mice. Conversely, the GluR1 protein level was maintained in old Pdyn KO mice, and the NMDA NR2B subunit level was increased by 42% when compared to old WT animals.
CONCLUSIONS:
These results suggest that elevated dynorphin expression occurring during aging and AD may mediate cognitive deficits by altering the glutamatergic system integrity.
http://www.ncbi.nlm....pubmed/23970097
The κ
opioid system regulates endothelial cell differentiation and pathfinding in vascular development.
http://www.ncbi.nlm....pubmed/21460241
Prodynorphin knockout mice demonstrate diminished age-associated impairment in spatial water maze performance.
Nguyen XV1,
Masse J,
Kumar A,
Vijitruth R,
Kulik C,
Liu M,
Choi DY,
Foster TC,
Usynin I,
Bakalkin G,
Bing G.
Abstract
Dynorphins, endogenous kappa-opioid agonists widely expressed in the central nervous system, have been reported to increase following diverse pathophysiological processes, including excitotoxicity, chronic inflammation, and traumatic injury. These peptides have been implicated in cognitive impairment, especially that associated with aging. To determine whether absence of dynorphin confers any beneficial effect on spatial learning and memory, knockout mice lacking the coding exons of the gene encoding its precursor prodynorphin (Pdyn) were tested in a water maze task. Learning and memory assessment using a 3-day water maze protocol demonstrated that aged Pdyn knockout mice (13-17 months) perform comparatively better than similarly aged wild-type (WT) mice, based on acquisition and retention probe trial indices. There was no genotype effect on performance in the cued version of the swim task nor on average swim speed, suggesting the observed genotype effects are likely attributable to differences in cognitive rather than motor function. Young (3-6 months) mice performed significantly better than aged mice, but in young mice, no genotype difference was observed. To investigate the relationship between aging and brain dynorphin expression in mice, we examined dynorphin peptide levels at varying ages in hippocampus and frontal cortex of WT 129SvEv mice. Quantitative radioimmunoassay demonstrated that dynorphin A levels in frontal cortex, but not hippocampus, of 12- and 24-month mice were significantly elevated compared to 3-month mice. Although the underlying mechanisms have yet to be elucidated, the results suggest that chronic increases in endogenous dynorphin expression with age, especially in frontal cortex, may adversely affect learning and memory.
http://www.ncbi.nlm....pubmed/15922052
Sorry if this seems like spam, but opioids yield a lot of nice results when used with cell death differentiation proliferation, stem cell control etc. In addition to all that they already do. And when you think about it analgesia is a trigger for changes. Ok, I'll numb it up for now, but changes need to be made for it to be ready better later, or triaged/severed.










































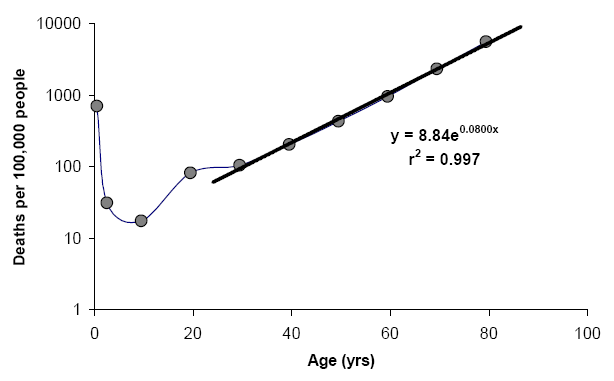




 ). So I was right, there were exactly 5 people under 25 and one ~27. The second graph has only 2 people <25 (the second looks exactly 25) and, as you can see, it shows decline of pentosidine with age for those two, which, however, proves nothing, just like the first graph proves nothing, 'cause the sample size is just too small.
). So I was right, there were exactly 5 people under 25 and one ~27. The second graph has only 2 people <25 (the second looks exactly 25) and, as you can see, it shows decline of pentosidine with age for those two, which, however, proves nothing, just like the first graph proves nothing, 'cause the sample size is just too small.
 not a good sign
not a good sign 














