.
S O U R C E : Frontiers in IMMUNOLOGY
Lack of immune system cells or impairment in differentiation of immune cells is the basis for many chronic diseases. Metabolic changes could be the root cause for this immune cell impairment. These changes could be a result of altered transcription, cytokine production from surrounding cells, and changes in metabolic pathways. Immunity and mitochondria are interlinked with each other. An important feature of mitochondria is it can regulate activation, differentiation, and survival of immune cells. In addition, it can also release signals such as mitochondrial DNA (mtDNA) and mitochondrial ROS (mtROS) to regulate transcription of immune cells. From current literature, we found that mitochondria can regulate immunity in different ways. First, alterations in metabolic pathways (TCA cycle, oxidative phosphorylation, and FAO) and mitochondria induced transcriptional changes can lead to entirely different outcomes in immune cells. For example, M1 macrophages exhibit a broken TCA cycle and have a pro-inflammatory role. By contrast, M2 macrophages undergo β-oxidation to produce anti-inflammatory responses. In addition, amino acid metabolism, especially arginine, glutamine, serine, glycine, and tryptophan, is critical for T cell differentiation and macrophage polarization. Second, mitochondria can activate the inflammatory response. For instance, mitochondrial antiviral signaling and NLRP3 can be activated by mitochondria. Third, mitochondrial mass and mobility can be influenced by fission and fusion. Fission and fusion can influence immune functions. Finally, mitochondria are placed near the endoplasmic reticulum (ER) in immune cells. Therefore, mitochondria and ER junction signaling can also influence immune cell metabolism. Mitochondrial machinery such as metabolic pathways, amino acid metabolism, antioxidant systems, mitochondrial dynamics, mtDNA, mitophagy, and mtROS are crucial for immune functions. Here, we have demonstrated how mitochondria coordinate to alter immune responses and how changes in mitochondrial machinery contribute to alterations in immune responses. A better understanding of the molecular components of mitochondria is necessary. This can help in the development of safe and effective immune therapy or prevention of chronic diseases. In this review, we have presented an updated prospective of the mitochondrial machinery that drives various immune responses.
Introduction
Mitochondria have many fundamental functions such as energy production, providing metabolites for building macromolecules, and aiding in differentiation, apoptosis, and cell cycle. After reviewing recent literature, two mitochondrial functions appeared to be intriguing. First, mitochondria and the endoplasmic reticulum (ER) communicate with each other through signaling molecules (1). Second, mitochondria are associated with NLRP3 inflammasome activation (2). Mitochondria are localized near the ER to supply energy for protein and lipid synthesis. For example, Bantug et al. have highlighted that ER and mitochondria junction signaling is critical in CD8+ memory T cells. Mechanistically, this happens in crosstalk between ER and mitochondria by mammalian target of rapamycin complex 2, AKT (protein kinase B), and glycogen synthase kinase 3β mediated signaling which promotes respiration (Figure 1.9) (3).
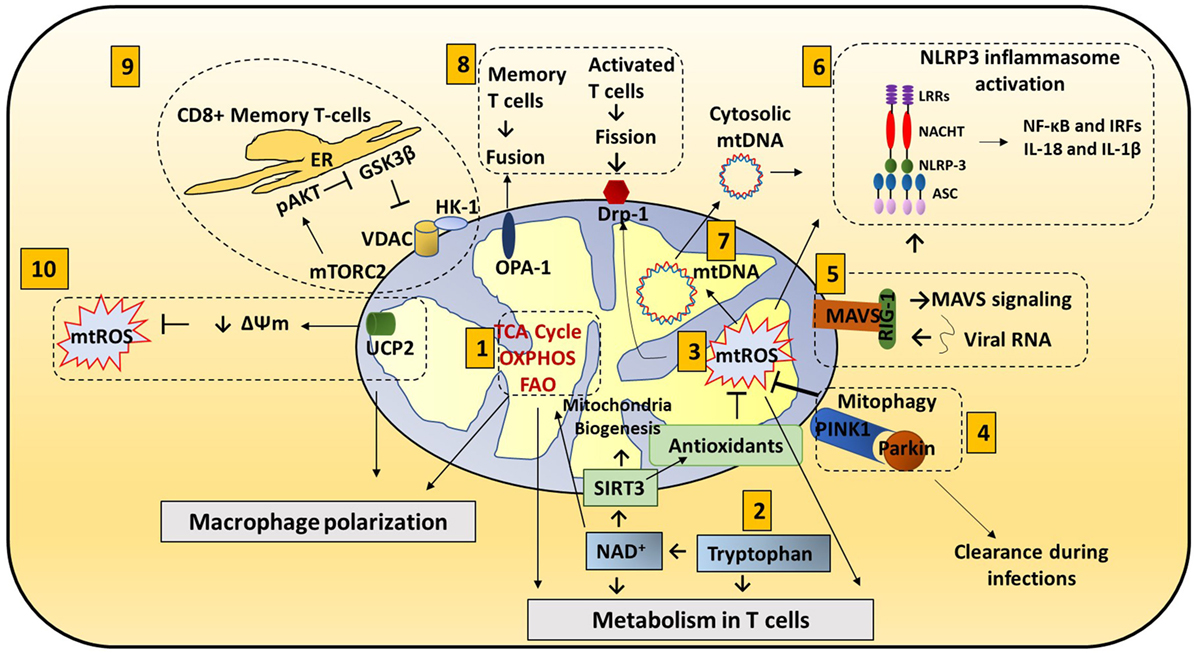
Figure 1. Mitochondria roles in immune responses: mitochondrial components are involved in immune functions. (1) Metabolic pathways such as TCA cycle, oxidative phosphorylation (OXPHOS), and fatty acid oxidation (FAO) are important for macrophage polarization and T cell differentiation. (2) Amino acid metabolism (Tryptophan metabolism is shown here.) also contributes to mitochondrial immune functions. Tryptophan depletion can cause immune tolerance. NAD+ is synthesized from tryptophan. NAD+ contributes to SIRT3-mediated mitochondrial biogenesis. In addition, NAD+ is required for metabolic pathways and CD4+ T cell differentiation. (3) Mitochondrial ROS (mtROS) control immune cell transcription, metabolism, and NLRP3 mediated inflammation. Antioxidants such as glutathione balance mtROS. SIRT3 is involved in regulating many mitochondrial proteins such as IDH2 (TCA cycle), MnSOD [reactive oxygen species (ROS) balance], and FOXO3. SIRT3 inhibits mtROS. (4) Mitophagy is crucial for removing damaged mitochondria. Damaged mitochondria are a source of mtROS so mitophagy balances mtROS. Pink1, localized on the mitochondrial outer membrane, binds to parkin and initiates mitophagy. Parkin mutations can increase the susceptibility to the intracellular bacteria Mycobacterium leprae and Salmonella enterica. Decreased mitophagy results in increased ROS which further increases the susceptibility to infections. Hepatitis B and C viruses use mitophagy to their benefit. These viruses protect themselves from mitochondria induced apoptosis by activating mitophagy. (5) Mitochondrial antiviral signaling (MAVS) in the mitochondria membrane initiates the inflammatory response. Upon viral infections, viral RNA is sensed by RIG-I. RIG-I activates MAVS (located on the outer membrane of mitochondria). MAVS signaling can activate NLRP3 inflammasome and NF-κB/IRF transcription. (6) NLRP3 inflammasome contributes to the activation of caspase-1 and leads to NF-κB signaling and production of IL-1β and IL-18. (7) Mitochondrial DNA (mtDNA) can be released from mitochondria into the cytosol and activate the NLRP3 inflammasome and production of IL-1β and IL-18. Also, mtROS can induce mtDNA mutation. (8) Mitochondrial dynamics, fission (OPA-1) and fusion (Drp1), is associated with activated T cells and memory T cells, respectively. (9) ER and mitochondria junction: in CD8+ memory T cells, mitochondria are placed near the ER. HK-1 (hexokinase-1, an important enzyme in glycolysis) in conjunction with pAKT, mammalian target of rapamycin complex 2 (mTORC2), and glycogen synthase kinase 3β (GSK-3β) mediated signaling cascades regulate CD8+ memory T cell metabolism. (10) Uncoupling protein 2 (UCP2), a mitochondrial membrane protein, allows protons to enter the mitochondrial matrix, thereby decreasing mitochondrial membrane potential which further decreases mtROS.
Immune response and metabolism are closely dependent on each other. During immune response, immune cells transition from metabolic quiescence to active phase. This transition is associated with a metabolic shift from catabolic to anabolic state. During the quiescence state, macromolecules undergo catabolic pathways to produce energy and support long-term survival. During the anabolic state, macromolecules are synthesized and support a balance between the need for ATP and required metabolites. Depending on the need, immune cells choose a specific pathway such as β-oxidation to generate more ATP than glycolysis. Thus, ATP and metabolic intermediates provide the signals to activate immune responses (4–6).
Hence, mitochondria, the chief organelle for metabolism, have emerged to play crucial roles in the maintenance and establishment of immune responses. Mitochondrial machinery such as metabolic pathways, amino acid metabolism, antioxidant systems, mitochondrial dynamics, mitochondrial DNA (mtDNA), mitophagy, and mitochondrial ROS (mtROS) are crucial for immune functions. In this review, we have presented an updated prospective of the mitochondrial machinery that drives various immune responses.
Metabolic Pathways are Tightly Controlled in Immune Cells
Mitochondrial metabolic events can have tremendous impact on immune cell function. These metabolic events could be guided by the internal signals from the cell itself or influenced by other surrounding cells. Here, we have pointed out how oxidative phosphorylation (OXPHOS), fatty acid metabolism, and amino acid metabolism in mitochondria influence immune cell activity (Figure 1.1).
OXPHOS Affects Immune Cell Activity M1 and M2 Polarization
There are two categories of macrophages—M1 (classically activated) and M2 (alternatively activated). Polarization of monocytes to M1 or M2 phenotypes is controlled by the cytokines produced by other immune cells. M1 macrophages are activated by IFN-γ produced by Th1 cells and lipopolysaccharide (LPS). Plus, M1 exhibits nitric oxide (NO) production (7) and a pro-inflammatory phenotype. Contrarily, M2 macrophages are activated by IL-4 or IL-13 to regulate anti-inflammation and promote Th2 response and tissue repair (Figure 2A) (8).
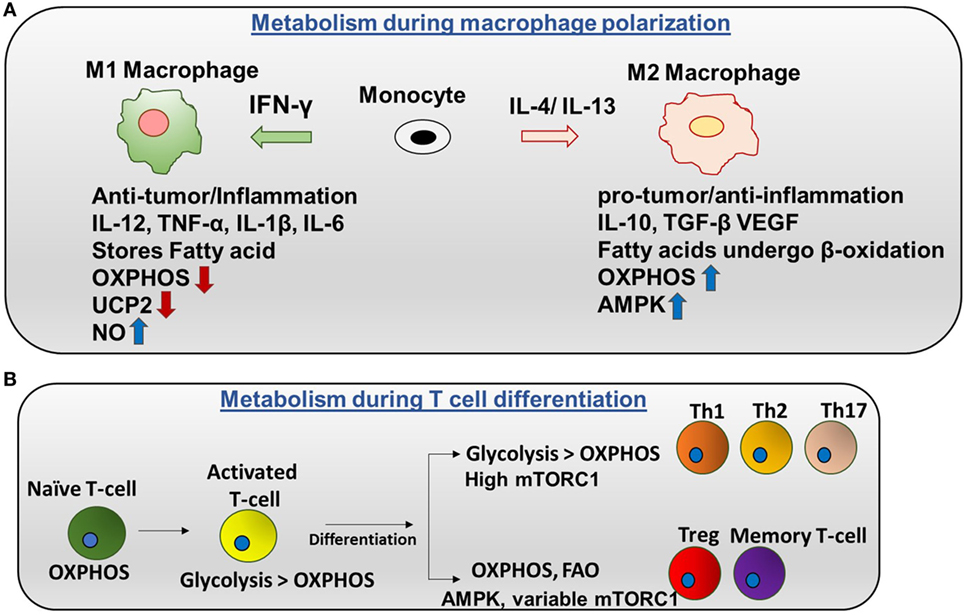
Figure 2. Metabolic regulation in macrophages and T cells: (A) metabolism during macrophage polarization: polarization of monocytes to M1 or M2 phenotypes is controlled by the cytokines produced by other immune cells. M1 macrophages are activated by IFN-γ produced by Th1 cells. M1 macrophages tend to store surplus FA as triacyclglycerols and cholesteryl esters in lipid droplets, and they exhibit higher aerobic glycolysis and lower oxidative phosphorylation (OXPHOS). Nitric oxide production is higher in M1. Uncoupling protein 2 (UCP2) expression is decreased in M1 macrophages. Contrarily, M2 macrophages are activated by IL-4 or IL-13 to regulate anti-inflammation and promote Th2 response and tissue repair. M2 macrophages adopt a metabolic program dominated by fatty acid-fueled OXPHOS and channel FA toward re-esterification and β-oxidation. Silencing UCP2 impairs M2 macrophage activation by IL-4. High adenosine monophosphate-activated protein kinase (AMPK) and low NO is the reason for high OXPHOS in M2 macrophages. (B) Metabolism during T cells differentiation: naïve T cells are dependent on OXPHOS as their primary metabolic pathway. By contrast, activated T cells exhibit higher glycolysis than OXPHOS. After differentiation, Th1, Th2, and Th17 have higher glycolysis than OXPHOS and high mTORC1 activity. Memory T cells and regulatory T cells undergo AMPK-dependent FAO and have variable mTORC1.
Uncoupling Protein 2 (UCP2) and Macrophage Polarization
Mitochondrial UCP2 is localized in the mitochondrial inner membrane and shuttle protons toward the matrix (Figure 1.10). There is increasing evidence supporting that UCP2 controls mitochondria derived reactive oxygen species (ROS). UCP2 can also influence polarization of macrophages. UCP2 expression is decreased in M1 macrophages. By blocking UCP2, there is a decrease in IL-4 induced M2 macrophage activation (9). However, how UCP2 is regulated in other immune cells is not well elucidated.
TCA Cycle in M1 Macrophages
Metabolic events are tightly controlled in M1 and M2 macrophages. Mechanistically in M1 macrophages, TCA (tricarboxylic acid) cycle exhibits two breaks (Figure 3A) (10, 11). First break occurs in the enzymatic step involving isocitrate dehydrogenase (IDH). This results in increased citrate and itaconic acid levels. Citrate is the precursor for fatty acid (FA) synthesis, prostaglandin (PG), and nitric oxide (NO) production. Itaconic acid has anti-bacterial properties which supports the notion that M1 macrophages have inflammatory function. Interestingly, IDH1 and IDH2 are the enzymes that catalyze decarboxylation of isocitrate to α-ketoglutarate outside and inside of the mitochondria, respectively (12). IDH2 plays a vital role in the formation of NADPH which is critical for ROS balance in the mitochondria (13). Second break occurs in the enzymatic step involving succinate dehydrogenase. This causes an increase in the expression of succinate. Succinate stabilizes HIF-1α. HIF-1α binds to the IL-1β promoter and promotes IL-1β production. Increased aspartate arginosuccinate shunt will further increase the flow of the TCA cycle. Therefore, this will increase citrate levels and the urea cycle that contribute to NO production. Inhibition of aspartate aminotransferase inhibits NO and IL-6 in M1 macrophages. Thus, the production of NO, IL-1β, and itaconic acid can promote inflammatory functions (14). Also, glutamine metabolism also impacts TCA cycle in M1 macrophages.
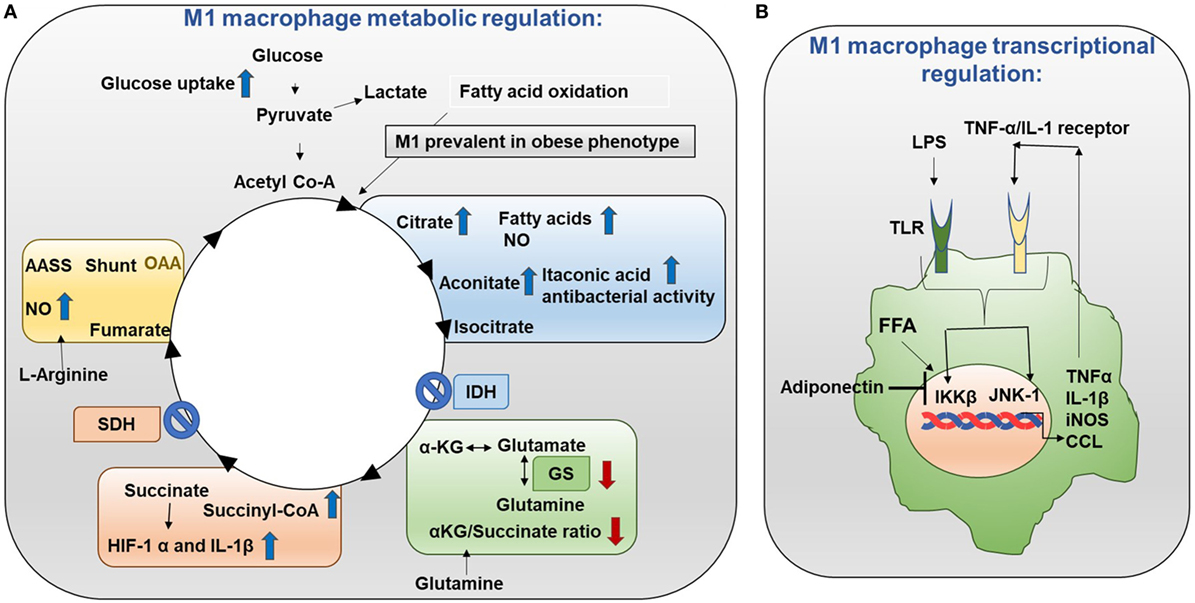
Figure 3. Metabolism in M1 macrophages. (A) M1 macrophage metabolic regulation: M1 macrophages are prevalent in obese adipose tissue. Glucose uptake is increased in M1 macrophages. Importantly, the TCA cycle exhibits two breaks. The first break involves the enzyme isocitrate dehydrogenase (IDH) which results in increased levels of citrate and itaconic acid. Citrate feeds fatty acid (FA) synthesis for prostaglandin (PG) and nitric oxide (NO) production while itaconic acid has anti-bacterial properties. The second break happens with the enzyme succinate dehydrogenase (SDH) which causes increased succinate levels. Succinate stabilizes HIF-1α which binds to the interleukin (IL)-1β promoter boosting IL-1β production and inflammation. Increased flow through the aspartate arginosuccinate shunt (AASS) replenishes the TCA cycle which further increases citrate levels and feeds the urea cycle which contributes to NO production. Glutamine is converted into glutamate by glutamate synthase (GS). Glutamate can further be converted into α-ketoglutarate (αKG). Low αKG/succinate ratio strengthens M1 macrophage activation. Glutamine-synthetase inhibition skews M2-polarized macrophages toward the M1-like phenotype characterized by reduced intracellular glutamine and increased succinate with enhanced glucose flux through glycolysis which is partly related to HIF-1α activation. (B) M1 macrophage transcriptional regulation: in M1 macrophages, the TCA cycle is broken. As a result, citrate is converted to free fatty acids (FFAs). FFAs activate macrophage IKK and JNK1 signaling molecules to induce M1 polarization. Activation of IKK and JNK1 in macrophages produces IL-1β and TNFα which can initiate inflammatory response. Adiponectin is released from adipose tissue and is involved in the breakdown of fatty acids. Adiponectin signaling inhibits M1 programming.
TCA Cycle in M2 Macrophages
M2 macrophages adopt a metabolic program fueled by fatty acids and OXPHOS. Low NO production and high adenosine monophosphate-activated protein kinase (AMPK) can be the reason for the high rate of OXPHOS in M2 macrophages (Figure 4A) (15). Knockdown of pyruvate dehydrogenase kinase 1 diminishes M1 activation whereas it enhances M2 activation of macrophages (16).
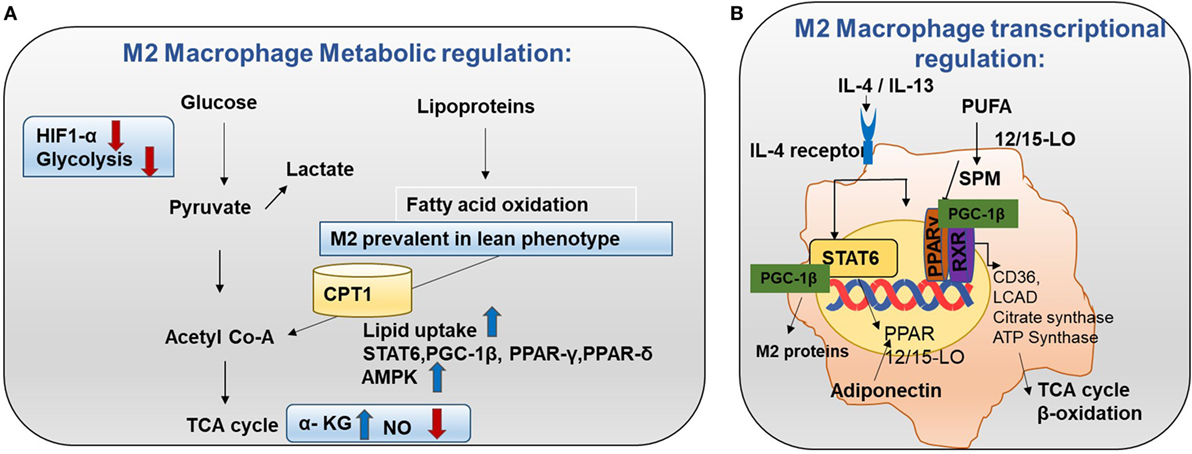
Figure 4. Metabolism in M2 macrophages. (A) M2 macrophage metabolic regulation: M2 macrophages adopt a metabolic program fueled by fatty acids and oxidative phosphorylation (OXPHOS). M2 macrophages are seen in lean adipose tissue and lipid uptake is increased in M2 macrophages (8). Both OXPHOS and FAO are important for the anti-inflammatory function of M2 macrophages. In M2 macrophages, glycolysis is impaired due to low HIF-1α activity and low expression of the active 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 1 (PFKFB1). Also, high α-KG/succinate ratio promotes the M2 phenotype. This could be due to the high flow of the TCA cycle or glutamine metabolism. M2 macrophages will channel FA toward re-esterification and β-oxidation. β-oxidation is channeled through carinitine palmitoyl transferase 1 (CPT1) (9). The high rates of OXPHOS in M2 macrophages are caused by low nitric oxide (NO) production and high adenosine monophosphate-activated protein kinase (AMPK) activity. (B) M2 macrophage transcriptional regulation: omega-3 polyunsaturated fatty acids (PUFA) undergo enzymatic transformation by 12/15-LO (lipoxygenase) and other enzymes in the production of specialized pro-resolving lipid mediators (SPMs) and intermediate metabolites that are potent PPAR ligands. M2-inducing cytokines IL-4 and IL-13 promote STAT6 phosphorylation to induce transcription of PPARs (γ and δ) and their coactivator PPARγ-coactivator-1β (PGC-1β). M2 exhibit enhanced PPAR-γ activity. PPAR-γ forms a heterodimer with RXR to induce OXHOS regulating proteins. PGC-1β promotes the STAT6 transcription complex and expression of M2 proteins such as arginase-1 and the pattern-recognition/endocytic receptor CD206. The STAT6 transcription complex further increases transcription of TCA cycle enzymes such as citrate synthase, CD36 (fatty acid transporter), ATP synthase, 12/15 lipoxygenase (converts omega-3 PUFA to SPMs), and LCAD (long chain acyl dehydrogenase, an enzyme for β-oxidation). Adiponectin activates PPARs in M2 macrophages and consequently increases β-oxidation.
.../...
F O R T H E R E S T O F T H E S T U D Y , P L E A S E V I S I T T H E S O U R C E