
































.
F U L L T E X T S O U R C E : BMC Genomics
Abbreviations
GWAS
Genome-Wide Association Study
NB
Naïve Bayes
POV
Percentage of Variance
PPHAGE
Prediction and Prioritization of Human Aging Genes
PUL
Positive-unlabeled learning
ROC
Receiver Operating Characteristic
Abstract
Background
Machine learning can effectively nominate novel genes for various research purposes in the laboratory. On a genome-wide scale, we implemented multiple databases and algorithms to predict and prioritize the human aging genes (PPHAGE).
Results
We fused data from 11 databases, and used Naïve Bayes classifier and positive unlabeled learning (PUL) methods, NB, Spy, and Rocchio-SVM, to rank human genes in respect with their implication in aging. The PUL methods enabled us to identify a list of negative (non-aging) genes to use alongside the seed (known age-related) genes in the ranking process. Comparison of the PUL algorithms revealed that none of the methods for identifying a negative sample were advantageous over other methods, and their simultaneous use in a form of fusion was critical for obtaining optimal results (PPHAGE is publicly available at https://cbb.ut.ac.ir/pphage).
Conclusion
We predict and prioritize over 3,000 candidate age-related genes in human, based on significant ranking scores. The identified candidate genes are associated with pathways, ontologies, and diseases that are linked to aging, such as cancer and diabetes. Our data offer a platform for future experimental research on the genetic and biological aspects of aging. Additionally, we demonstrate that fusion of PUL methods and data sources can be successfully used for aging and disease candidate gene prioritization.
Background
Prior understanding of the genetic basis of a disease is a crucial step for the better diagnosis and treatment of the disease [1]. Machine learning methods help specialists and biologists the use of functional or inherent properties of genes in the selection of candidate genes [2]. Perhaps the question that is posed to researchers is why all research is aimed at identifying pathogenic rather than non-pathogenic genes. The answer may lie in the fact that genes introduced as non-pathogens may be documented as disease genes later on.
Biologists apply computation, mathematics methods, and algorithms to develop machine learning methods of identifying novel candidate disease genes [3]. Based on the principle of “guilt by association”, similar or identical diseases share genes that are very similar in function or intrinsic properties, or have direct physical protein-protein interactions [4]. Most methods of predicting candidate genes employ various biological data, such as protein sequence, functional annotation, gene expression, protein-protein interaction networks, regulatory data and even orthogonal and conservation data, to identify similarities with respect to the principle of association based on similarity [5]. These methods are categorized as unsupervised, supervised, and semi-supervised [6]. Unsupervised methods cluster the genes based on their proximity and similarity to the known disease genes, and rank them by various methods. Supervised methods create a boundary between disease genes and non-disease genes, and utilize this boundary to select candidate genes. Several studies have been performed to address different aspects of the methodology and have expanded the use of various methods and tools [3, 7, 8, 9, 10, 11, 12].
The tools that are available for candidate gene prioritization can be classified with respect to efficiency, computational algorithms, data sources, and availability [13, 14, 15]. Available prioritization tools can be categorized into specific and general tools [16]. Specific tools are used to prioritize candidate genes associated with a specific disease. In these methods, information related to a specific tissue involved in the disease or other information related to the disease is employed. General tools can be applied for most diseases, and various data sources are often used in these tools. Gene prioritization tools can be divided into two types of single-species and multi-species. Single-species tools are only usable for a specific species, such as human or mouse. Multi-species tools have the ability to prioritize candidate genes in several different species. For example, the ENDEAVOR software can prioritize the candidate genes in six different species [17]. With respect to computational algorithms, candidate prioritization tools are primarily divided into two groups of complex network-based methods and similarity-based methods [5]. The inevitable completeness and existence of errors in biological data sources necessitate fusion of multiple data sources [18]. Most gene targeting methods, therefore, use multiple data sources to improve performance.
The purpose of this study was to design a machine to identify and prioritize novel candidate aging genes in human. We examined the existing methods of identifying human non-aging (negative) genes in the machine learning techniques, and then made a binary classifier for predicting novel candidate genes, based on the positively and negatively learned genes. Gene ranking was based on the principle of the similarity among positive genes through “guilt by association”. Thus, across the unlabeled genes, genes that were less similar in respect with the known genes were employed as negative sample.
Results
The three positive unlabeled learning (PUL) algorithms, Naïve Bayes (NB), Spy, and Rocchio-SVM, were used to evaluate the underlying data, and to compare them to the eight datasets introduced with respect to performance. All samples of a class with a higher frequency were unlabeled. We applied the algorithm to predict the labels. These methods utilize a two-step strategy and are intended to extract a reliable negative sample from the main data (Table 1).
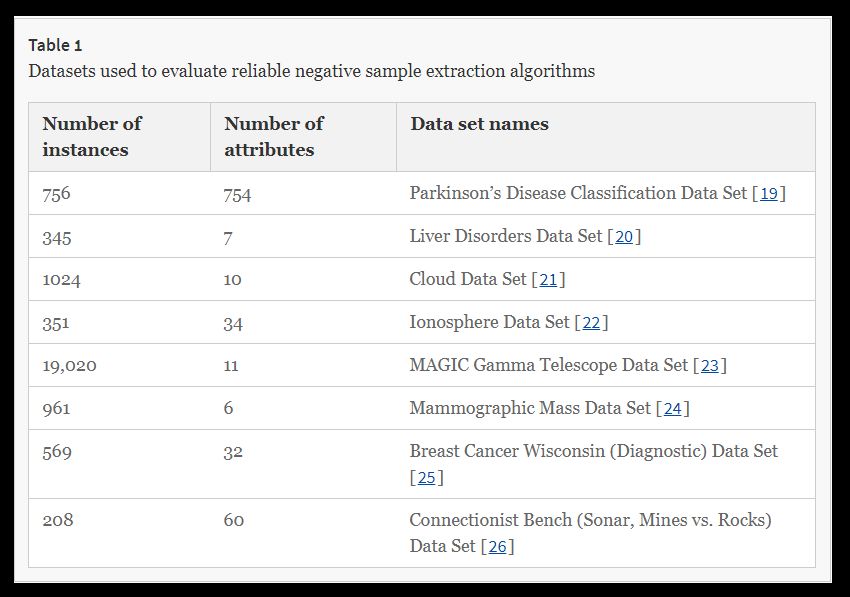
We also randomly selected 70% of the positive samples as the training set, and the remainder as the test set. To determine the classifier, positive and negative samples were equally selected to ensure that the classifier did not have any bias at the training step. Therefore, we compared the three algorithms with eight data sources extracted from the UCI database (Additional file 1).
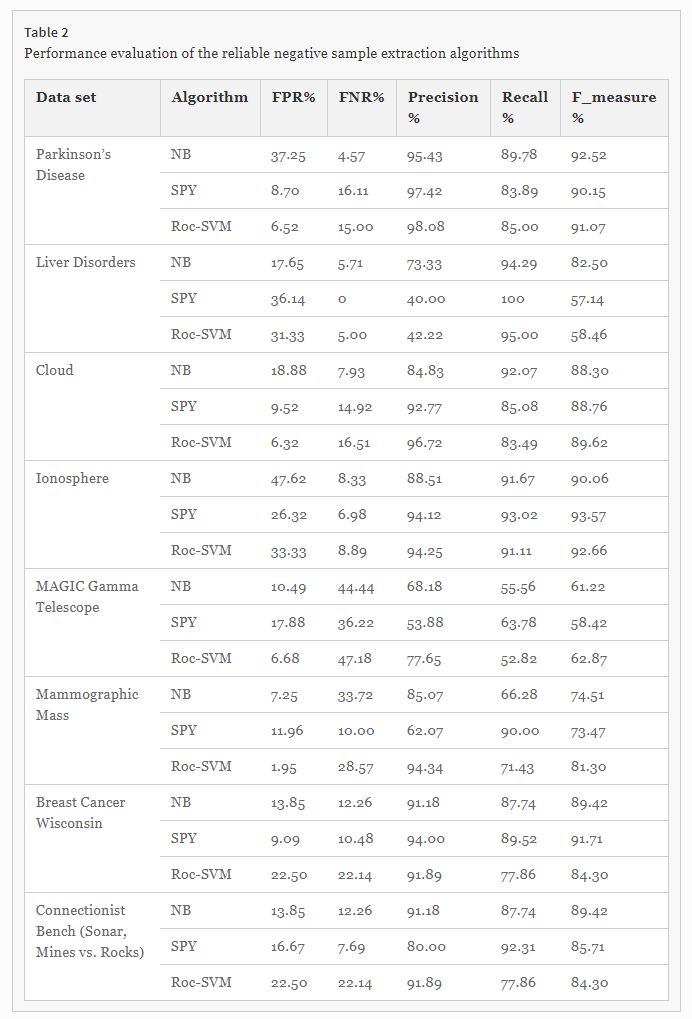
Comparison of the parameters of the three algorithms for all data sets revealed similar results in F_measure. For example, in data set 1, the precision of the Roc-SVM method, (approximately 2–3%,) was better than those of the other two methods. However, the recall of the NB method (approximately 4–6%,) was better than those of the other two methods, and Roc-SVM method had a lower false positive rate than that of the other two methods (Table 2). In addition, comparison between the parameters of the three algorithms for data set 2, revealed that the precision of the NB method was better than that of the other two methods, the recall SPY method was 5% better than that of the other two methods, and the NB method had a lower false positive rate than that of the other two methods. Therefore, none of the methods had an absolute superiority. Since the results were very similar, the output of the three methods was combined.
.../...
.
Edited by Engadin, 14 November 2019 - 07:21 PM.













