
































.
F U L L T E X T S O U R C E : AgING
Abstract
DNA methylation changes within the genome can be used to predict human age. However, the existing biological age prediction models based on DNA methylation are predominantly adult-oriented. We established a methylation-based age prediction model for children (9-212 months old) using data from 716 blood samples in 11 DNA methylation datasets. Our elastic net model includes 111 CpG sites, mostly in genes associated with development and aging. The model performed well and exhibited high precision, yielding a 98% correlation between the DNA methylation age and the chronological age, with an error of only 6.7 months. When we used the model to assess age acceleration in children based on their methylation data, we observed the following: first, the aging rate appears to be fastest in mid-childhood, and this acceleration is more pronounced in autistic children; second, lead exposure early in life increases the aging rate in boys, but not in girls; third, short-term recombinant human growth hormone treatment has little effect on the aging rate of children. Our child-specific methylation-based age prediction model can effectively detect epigenetic changes and health imbalances early in life. This may thus be a useful model for future studies of epigenetic interventions for age-related diseases.
Introduction
Due to the declining fertility rate and the increasing life expectancy, the world is on the brink of a demographic milestone: adults above the age of 65 will soon outnumber children under the age of 5 [1–3]. Population aging is accompanied by increases in illness, disability and dependency [4]. Consequently, noncommunicable diseases that more commonly occur in adults and older people are imposing the greatest burden on global health. Extending the period of life free of disability and disease is the key to limiting health and social costs. The most promising approach to this end is to identify age-related biological changes in body function or structure that are more accurate than the chronological age in predicting the future onset of age-related diseases or the remaining years of life [5].
Biomarkers of biological aging can be classified as molecular markers (based on DNA, RNA, etc.) or phenotypic biomarkers (based on anthropometric data such as bone age assessment, blood pressure, lipid levels, etc.) [5]. However, most studies of such biomarkers have been conducted in animals or older individuals [6–9]. Animals with short lifespans are typically not accurate models of the complex multifactorial exposures during human aging. Furthermore, most elderly study participants already suffer from age-related diseases. The theory of the fetal origins of adult disorders [10–13] proposes that many health problems in adults or the elderly are rooted in early life experiences and living conditions. Thus, interventions to reverse or delay age-related diseases and aging itself must be performed in childhood. The lack of tools to quantify aging in children is a significant obstacle to this goal.
Thus far, the most remarkable biological age predictor has been the epigenetic clock. Hannum et al. built a quantitative model of aging by measuring over 450,000 CpG markers in whole blood samples from 656 human subjects aged 19 to 101 years [14]. Horvath et al. developed a biomarker of aging called the multi-tissue predictor based on DNA methylation levels [15]. Using only three CpG sites, Weidner et al. constructed an age prediction model that was more precise than techniques based on telomere length [16]. The above studies demonstrated the feasibility of biological age prediction based on DNA methylation, but these models were mostly focused on adults. Although some of these predictive models included samples from children, the large age range (0–101 years old) and age unit (years) of these models reduced their accuracy both in predicting the biological ages of children (0 - 18 years old) and in revealing biologically relevant epigenetic abnormalities.
There has been some progress in DNA methylation research in children, but there are still many problems to be solved. For instance, Alisch et al. found 2078 age-associated CpG sites in boys (3–17 years old), but did not propose operational quantitative tools [17]. Almstrup et al. only used the data from 51 healthy children (5–16 years old) before and after pubertal onset to predict adolescent development [18]. Freire-Aradas et al. built a preliminary age prediction model using a dataset of 180 donors (2–18 years old) with the EpiTYPER® DNA methylation analysis system [19]. None of these studies completely covered the age range from 0 to 18 years, and each study used a single sample data type with a small number of samples, so the results were not highly accurate or applicable.
To delineate the aging pattern precisely throughout childhood, we analyzed DNA methylation datasets from a large cohort of children to construct a child-specific methylation-based age prediction model covering the whole age period from 0 to 18 years with a small age unit (months). Our model is a new tool for quantifying health imbalances and monitoring predictors of age-related diseases early in life, and thus may facilitate early prevention and intervention.
Results
Establishment of a child-specific methylation-based age prediction model
Characteristics of the DNA methylation datasets
We obtained publicly available DNA methylation datasets that were generated with the Illumina 27K or Illumina 450K array platform. Data from 716 healthy children aged 9 to 212 months from 11 different datasets were used to build the quantitative model (Table 1, Figure 1A). These healthy children included 529 boys and 187 girls (Supplementary Figure 1A). Nearly half of the samples (46.5%) were assessed on the Illumina 450K platform (Supplementary Figure 1B). We only studied the 21,979 CpG sites that were present on both Illumina platforms. For simplicity and accuracy, we discarded markers on sex chromosomes and markers with more than 10 missing values across the datasets. DNA methylation levels were recorded as β values between 0 (completely unmethylated) and 1 (completely methylated). To study the link between disease and methylation age in childhood, we also analyzed the datasets of children with diseases (Supplementary Table 1). Details on the above datasets and the data preprocessing steps are provided in the Materials and Methods.
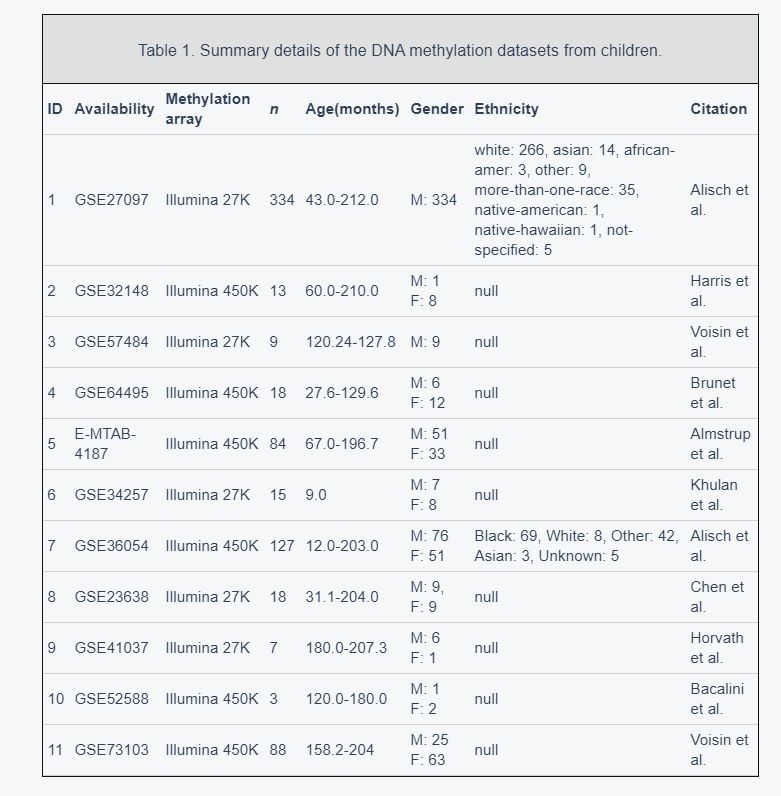
Table 1.
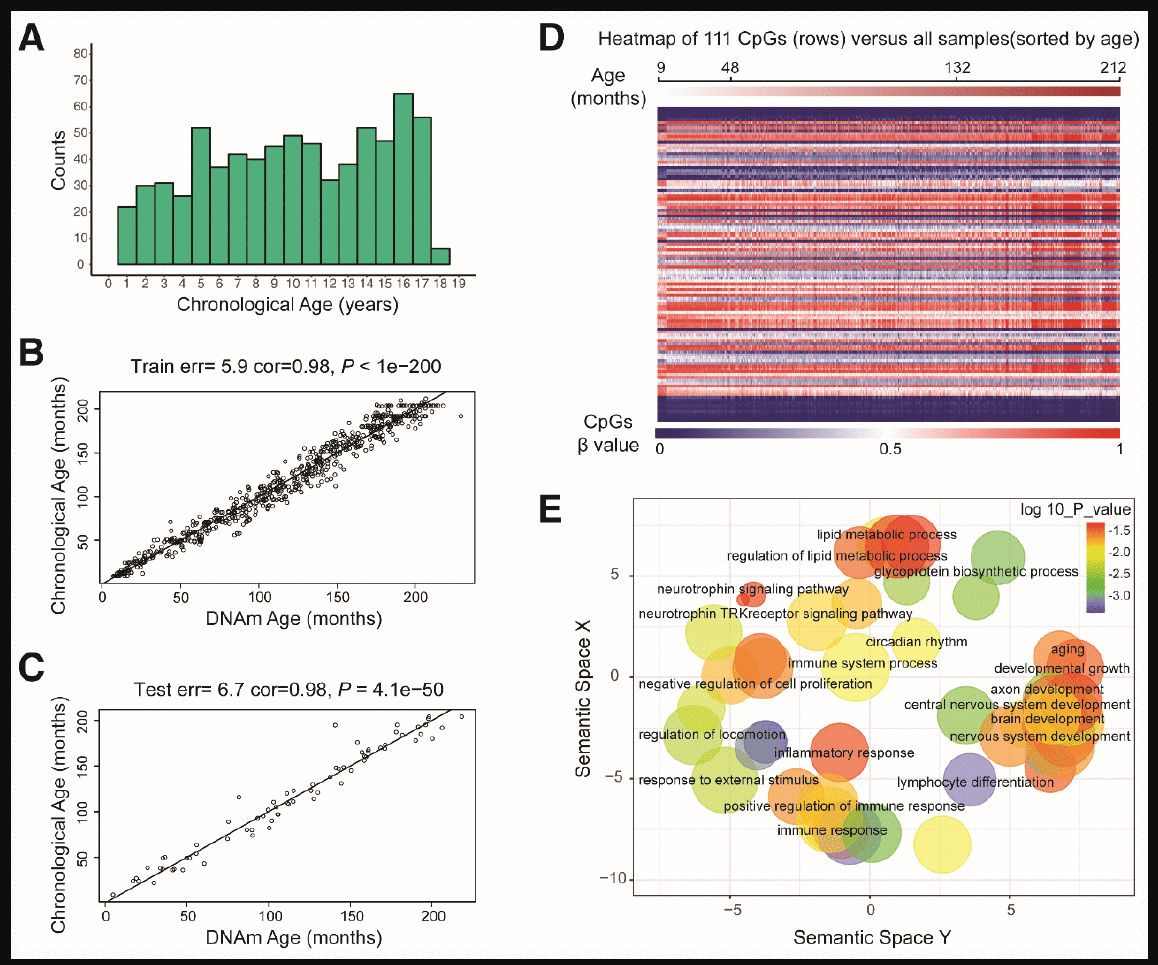
Figure 1. Characteristics of the prediction model. (A) Histogram of the age distribution for healthy children. The x-axis represents the chronological age of the individuals (age unit is years) and the y-axis (counts) represents the number of individuals. (B) Scatterplot of the DNA methylation (DNAm) age (x-axis) against the chronological age (y-axis) for the individuals in the training sets (age unit is months). For the training data, the correlation between the DNAm age and chronological age was 0.98, and the error (median absolute difference) was 5.9 months. © Scatterplot of the DNAm age (x-axis) against the chronological age (y-axis) for individuals in the test sets (age unit is months). For the test data, the correlation was 0.98 and the error was 6.7 months. (D) Heatmap of the DNA methylation levels of 111 CpG sites. Each row represents one CpG site, and the blue to red color spectrum represents β values from 0 to 1. The individuals are sorted by age (9 to 212 months), and it can be seen that the DNA methylation levels change with age. (E) Gene ontology analysis of the 111 CpG sites revealed several ontologies (P < 0.05) that may be associated with development and aging. Biological process gene ontologies were plotted in a sematic space with REVIGO, which groups related ontologies together.
A precise DNA methylation age prediction model in children
By combining sure independence screening [20] and penalized multivariate (elastic net) regression [21], we established a child-specific methylation-based age prediction model in the training cohort, and called the prediction value the DNA methylation age (Figure 2). K-fold cross-validation (k = 10) [22] was implemented to divide the training sets and test sets. The optimal model included 111 CpG sites that were accurately predictive of age (regression coefficients in Supplementary Table 2). In the training sets, the model was highly accurate, with a 98% correlation between age and predicted age, and an error of 5.9 months (Figure 1B). The accuracy remained the same when this model was validated on the test sets, as there was a 98% correlation between age and predicted age, and the error was 6.7 months (Figure 1C). The β values of the 111 CpG sites exhibited some certain trends with increasing age, although most of these changes were not very dramatic. The effects of age on the 111 CpG sites were visualized on a heat map, which showed the trends in DNA methylation across subjects (Figure 1D).
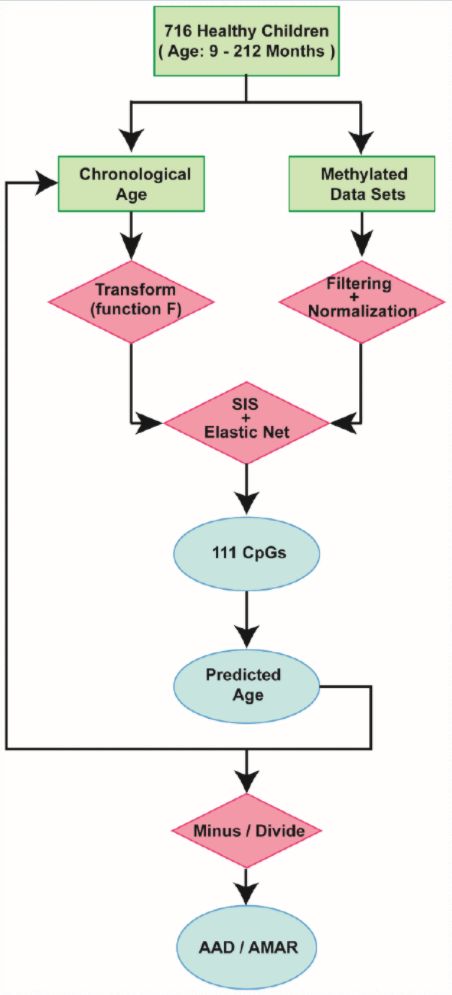
Figure 2. Schematic of the prediction model. A flow diagram of the child-specific methylation-based age prediction model. The green boxes represent the input data, the red diamonds represent the analysis methods and the blue ovals represent the prediction results. AAD: age acceleration difference; AMAR: apparent methylation aging rate; SIS: sure independence screening.
Age-related CpG sites associated with development and aging
To determine the biological functions of the 111 age-related CpG sites, we searched for significantly enriched GO terms (biological processes, cellular components and molecular functions, P < 0.05) and KEGG signaling pathways among the genes associated with these CpG sites. The top 20 GO terms and KEGG pathways are listed in Supplementary Figure 1. The GO terms were then drawn in a semantic space, and similar terms were combined. The results revealed clusters associated with developmental growth, immune responses, metabolic regulation and age-related diseases such as systemic lupus erythematosus, rheumatoid arthritis and cancer (Figure 1E, Supplementary Figure 1C and 1D, Supplementary Table 3).
Comparing the child-specific age predictor with other age predictors
We then explored the CpG sites selected for different age prediction models. There were only three overlapping sites among Hannum’s 71 CpGs [14], Horvath’s 353 CpGs [15] and our 111 CpGs (Figure 3Aa, b): cg04474832, cg09809672 and cg19722847. These sites are associated with the ABHD14A, EDARADD and IPO8 genes, respectively. Of the remaining 108 sites in our study, 59 (54.6%) overlapped with 2,078 previously-identified age-related sites in children [17]; these sites involved 43 genes (Figure 3Ac, d). We also compared our model with the model established by Freire-Aradas et al. [19], and obtained only two overlapping genes: EDARADD and PRKG2.

Figure 3. Comparison and verification of our model. (A) (a and b) Venn diagrams of the CpG sites (a) and genes associated with the CpG sites (b) selected from the three models. (c and d) Venn diagrams of the CpG sites © and genes (d) associated with age from our study and the study of Alisch et al. (B) Density plot of age and DNA methylation (DNAm) age. The red peak represents the chronological age, the green peak represents the DNAm age predicted by our model, and the blue peak represents the DNAm age predicted by Horvath et al. Dashed lines represent mean values. © Boxplot comparing the DNAm ages predicted by our model for monozygotic twins (paired t-test, n = 67 each for twins 1 and 2). The blue box indicates twin 1 and the yellow box indicates twin 2. (D) Boxplot comparing the DNAm ages predicted by the model of Horvath et al. for monozygotic twins (paired t-test, n = 67 each for twins 1 and 2). The blue box indicates twin 1 and the yellow box indicates twin 2. (E) Boxplot comparing the absolute values of the DNAm age differences of monozygotic twins predicted by the two models (two-sided t-test, n = 67). The blue box indicates the results from Horvath et al. and the yellow box indicates our results.
Since different data and methods were used to construct the above models, it was not possible to identify the most accurate model simply by comparing the error values of the models for the test sets. Thus, to further explore the accuracy and applicability of our model, we validated it with data from 67 pairs of monozygotic twins in the dataset GSE56105 [23]. We took this approach because the DNA methylation ages of healthy monozygotic twins who share the same genetic background and living environment should theoretically be similar. First, we used our model and the commonly used multi-tissue predictor [15] to calculate the DNA methylation ages of the monozygotic twins. The predictions from our model were closer to the actual ages of the twins, and the distribution of DNA methylation ages was more concentrated than that of the multi-tissue predictor (Figure 3B). Second, we compared the DNA methylation ages of twins 1 and 2 calculated by these two models. The predicted DNA methylation ages of twins 1 and 2 did not differ significantly when our model was used (Figure 3C, P = 0.51, paired t-test), while they did differ significantly when the multi-tissue predictor was used (Figure 3D, P = 0.025, paired t-test). Finally, we compared the absolute values of the DNA methylation age differences between twins 1 and 2 calculated by the two models. The absolute values of the two models differed significantly (Figure 3E, P < 0.01, t-test), and the values calculated by our model were closer to zero than those calculated by the multi-tissue predictor. Therefore, our prediction model performed better than the multi-tissue predictor in estimating children’s DNA methylation ages based on blood samples.
We could not compare the accuracy of our model with that of the model established by Freire-Aradas et al. [19] because the authors did not report their predicted age calculation formula and data; however, the error of 1.25 years (15 months) reported by Freire-Aradas et al. [19] was larger than the error of our model (6.7 months).
Aging patterns in children revealed by our model
Our aging model not only predicted the age of most children with high accuracy, but also revealed individual biological differences and aging trends in the pediatric population [14, 15]. To examine whether these differences were true biological differences (rather than measurement error or intrinsic variability), we used our aging model for two measurements of age acceleration. The first, called the age acceleration difference (AAD), is the DNA methylation age minus the chronological age. The second, called the apparent methylation aging rate (AMAR), is the DNA methylation age divided by the chronological age.
Age acceleration in children seems not to be influenced by gender or ethnicity
We then explored the association of the AAD and AMAR with the potentially clinically relevant factors of gender and ethnicity. In terms of gender, the mean AAD and AMAR values in all the healthy children’s samples were -0.01 months and 1.01, respectively. The AAD and AMAR values for boys were 0.003 months and 1.006, respectively, while the values for girls were -0.040 months and 1.020, respectively. Neither the AAD nor the AMAR differed significantly between boys and girls (AAD: P = 0.92, Wilcoxon test; Supplementary Figure 2A), although the AMAR was approximately 1.4% faster in girls than in boys (Supplementary Figure 2B). In contrast, in adults, the AMAR was reported to be 4% faster in men than in women [14]. This difference may be due to the fact that girls develop earlier than boys [24–26]. Regarding ethnicity, neither the AAD nor the AMAR differed significantly among children of different ethnicities (AAD: P = 0.9, AMAR: P = 0.26, ANOVA; Supplementary Figure 2C and 2D).
Age acceleration is the greatest in mid-childhood
We observed a trend in age acceleration in healthy children between the ages of 0 and 18 years. The age acceleration was close to zero before the age of 4, gradually rose after the age of 5, and fell to a negative value after the age of 12 (Figure 4A). To further explore the aging pattern in children, we divided childhood into three periods: toddlerhood (0–4 years), mid-childhood (5–11 years) and adolescence (12–18 years). We found that the AAD and AMAR were significantly greater in mid-childhood than in toddlerhood, and were significantly lower in adolescence than in mid-childhood (AAD: P = 7.2×10-14, AMAR: P = 4.5×10-6, ANOVA; Figure 4B and C). The same phenomenon was observed after sex stratification. Moreover, in mid-childhood, the aging rate seemed to be marginally faster in girls than in boys (Figure 4D, Supplementary Figure 3). These differences in the AAD and AMAR at different stages of childhood indicate that the aging rate of children is not completely consistent with the growth curve, which may be related to the development of several major organ systems.
.../...
.
















