
































.
F U L L T E X T S O U R C E : Clinical Epigenetics
Abstract
Background
The role of DNA methylation in aging has been widely studied. However, epigenetic mutations, here defined as aberrant methylation levels compared to the distribution in a population, are less understood. Hence, we investigated longitudinal accumulation of epigenetic mutations, using 994 blood samples collected at up to five time points from 375 individuals in old ages.
Results
We verified earlier cross-sectional evidence on the increase of epigenetic mutations with age, and identified important contributing factors including sex, CD19+ B cells, genetic background, cancer diagnosis, and technical artifacts. We further classified epigenetic mutations into High/Low Methylation Outliers (HMO/LMO) according to their changes in methylation, and specifically studied methylation sites (CpGs) that were prone to mutate (frequently mutated CpGs). We validated four epigenetically mutated CpGs using pyrosequencing in 93 samples. Furthermore, by using twins, we concluded that the age-related accumulation of epigenetic mutations was not related to genetic factors, hence driven by stochastic or environmental effects.
Conclusions
Here we conducted a comprehensive study of epigenetic mutation and highlighted its important role in aging process and cancer development.
Introduction
Epigenetic processes, among which DNA methylation is one of the most well studied, are fundamental in human aging [1]. Studies on DNA methylation have identified age-associated changes in methylation levels shared by individuals [2, 3], and have also reported an increasing divergence of methylation levels between individuals with age [4, 5].
Epigenetic mutations, defined as aberrant methylation levels that can lead to unusual gene expression, may be involved in cancer development and important for human aging [6, 7]. Unlike age-associated changes in methylation levels that are shared among individuals, the incidences of epigenetic mutations are rare, stochastic, and inconsistent between individuals. Recently, emerging studies on methylation variability have also identified differentially varied CpGs associated with cancer field defects [8, 9]. Epigenetic mutations can partly explain the increasing variability of methylation levels between individuals over time, but conversely, highly varied methylation sites do not necessarily contain extreme outliers. The extreme methylation levels may concur stronger biological consequences, such as cancer. Epigenetic mutations could contribute to the aging process through the accumulation of abnormally methylated CpGs (cytosine-phosphatase-guanine sites), which could further cause abnormal gene expression and downstream effects in tissues. A previous study by Gentilini et al [7] specifically defined epigenetic mutations as extreme outliers within a population, with methylation levels exceeding three times interquartile ranges (IQR) of the first quartile (Q1-3 × IQR) or the third quartile (Q3 + 3 × IQR). They found that the total numbers of epigenetic mutations increased exponentially with age. Also, studies using a similar outlier definition have identified methylation outlier in association with undesirable birth outcomes [10] and cancer [11]. However, the study on epigenetic mutations and aging was based on a cross-sectional study, it needs to be validated in a longitudinal setting, where the accumulation of epigenetic mutations over time can be followed within the same individuals. Moreover, it is not yet known what the clinical consequences of accumulated epigenetic mutations are, and if individuals with a high burden of epigenetic mutations are prone to develop cancer as previously suggested [6, 12].
In this study, we used a Swedish twin cohort including 375 individuals sampled up to five times in late life across 18 years (Table 1). We first validated the age-related increase of epigenetic mutations from a longitudinal perspective. Next, we identified important factors associated with the number of epigenetic mutations, including sex, cellular composition (CD19 B cells), genetic background, and technical artifacts. In parallel, we analyzed the direction of change in methylation level and characterized the epigenetic mutations as High- (HMO) and Low Methylation Outliers (LMO). We also studied the association between epigenetic mutations and cancer, as well as the genetic influence on epigenetic mutations using a twin approach. Last, we validated a select set of epigenetic mutations using bisulfite pyrosequencing.
Results
Longitudinal accumulation of epigenetic mutations is exponentially associated with age
To explore the longitudinal increase in number of epigenetic mutations, we measured DNA methylation data (Illumina 450k array) repeatedly in whole blood samples (n = 994) from participants in the Swedish Adoption/Twin Study of Aging (SATSA; Table 1) [13]. To avoid confounding by underlying genetic variation, we removed 20,660 CpGs that were associated with at least one single nucleotide polymorphism (SNP) (p < 1e-14) within 1 Mbps (mega base pairs), i.e., cis-methylation quantitative loci (cis-meQTLs). In the remaining 370,234 CpGs, the number of epigenetic mutations ranged from 58 to 26,291 in each sample, using the definition in Gentilini et al [7]. Across samples, the number of epigenetic mutations had a right-skewed distribution, which was close to normal distribution after log10-transformation (Additional file 1: Figure S1).
After identifying epigenetic mutations in SATSA, we found that the log10 total number of epigenetic mutations increased with age (p = 1.22e-13) longitudinally (Fig. 1a). We also identified additional factors and confounders associated with the number of epigenetic mutations (Table 2). Women had a slightly higher average number of epigenetic mutations than men (p = 6.33e-3). Low sample quality, as defined by the log10-transformed number of CpGs with detection p values over 0.01, was positively associated with the total number of epigenetic mutations (p = 1.48e-117). In general, unreliable samples tended to have more epigenetic mutations, indicating that measurement errors could also be identified as epigenetic mutations. However, after adjusting the mixed models for detection p value, the effect of age on number of epigenetic mutations remained unchanged. Using predicted cellular compositions, CD19+ B cell composition was positively associated with the total number of epigenetic mutations (p = 5.06e-23). After removing cis-meQTLs, the first genetic principal component (PC) showed only a minor effect on the total number of epigenetic mutation (p = 0.041).
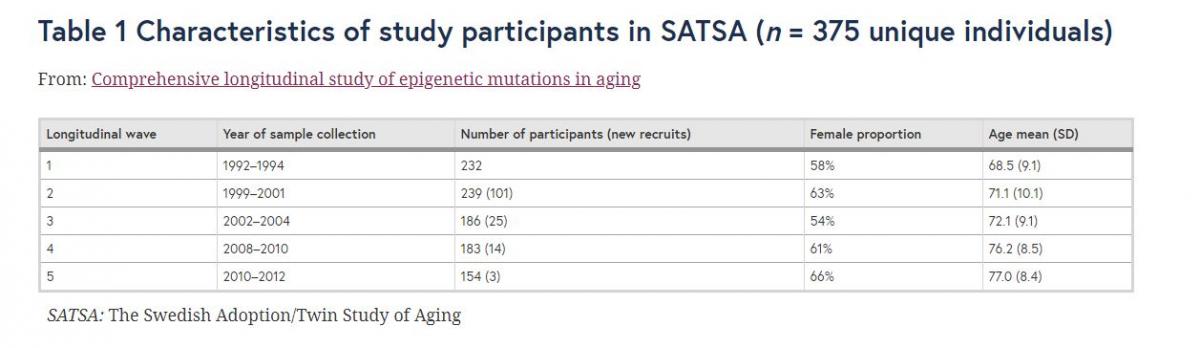
Fig. 1
The number of epigenetic mutations (log10-transformed) increased longitudinally with age in a longitudinal perspective using genome-wide DNA methylation data from repeated whole blood samples collected in the Swedish Adoption/Twin Study of Aging (SATSA; n = 375 participants). The numbers of epigenetic mutations of samples were counted from a total epigenetic mutations (n = 370,234 CpGs, p = 1.22e-13 for association with age), b total epigenetic mutation in individuals with at least 4 measures (p = 1.94e-08 for association with age), c frequent high methylation outliers (HMOs) (n = 969 CpGs, p = 2.09e-17 for association with age), d frequent HMOs in individuals with at least 4 measures (p = 1.07e-10 for association with age), e frequent low methylation outliers (LMOs) (n = 216 CpGs, p = 1.14e-05 for association with age), and f frequent LMOs in individuals with at least 4 measures (p = 5.76e-03 for association with age).
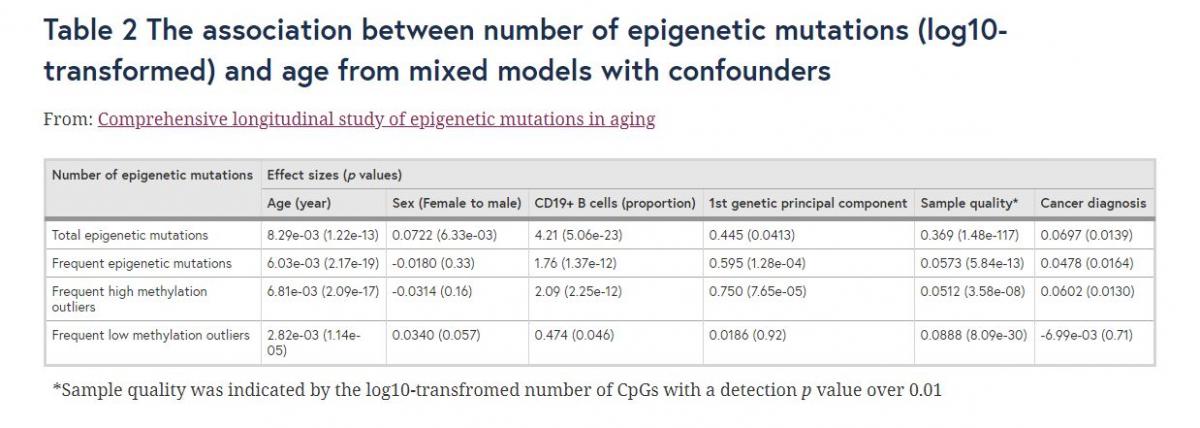
Out of all CpGs, 237,398 (64%) were defined as epigenetic mutations in at least one sample, but only 1,185 (0.32%) CpGs were mutated in more than 50 samples (5% samples), subsequently defined as frequently mutated CpGs. Only two of the 1,185 frequently mutated CpGs were also identified to be age-differentially methylated sites (aDMS) in our previous study [3]. The frequently mutated CpGs were still significantly associated with age, sample quality, CD19+ B cell compositions, and genetic PC1, while sex was no longer significant (Table 2).
High/low methylation outliers
Compared to normal methylation levels in the population, epigenetic mutations can be either higher or lower in methylation level. Hence, we defined HMO and LMO as CpGs with abnormally
higher or lower methylation levels than the average (Additional file 1: Figure S2). Of the defined epigenetic mutation sites, almost half were identified as HMOs and the other half as LMOs (118,259 HMOs and 119,175 LMOs). Thirty-six CpGs were defined as both HMOs and LMOs because those sites had intermediate methylation levels and very small IQRs. However, among the frequently mutated CpGs, there were significantly more HMOs than LMOs (969 and 216, p < 1e-16) (Fig. 2). Similar to the results of epigenetic mutations, both HMOs and LMOs were significantly associated with age (p = 1.98e-12 for HMOs and p = 1.73e-14 for LMOs), sex (p = 1.81e-3 for HMOs and p = 0.037 for LMOs), and B cells (p = 3.76e-22 for HMOs and p = 3.03e-20 for LMOs). Nevertheless, numbers of both sets of frequent mutations (log10-transformed) significantly increased with age (p = 2.09e-17 for HMOs and p = 1.14e-05 for LMOs) (Fig. 1c, e). Sex was no longer a significant factor with either frequent HMOs or LMOs. The composition of CD19+ B cell was still strongly associated with HMOs (p = 2.25e-12), but only marginally significant for LMOs (p = 0.046). Sample quality, as measured by detection p value, showed strong effects on both frequent HMOs and LMOs; however, LMOs were much more influenced (p = 8.09e-30) than HMOs (p = 3.58e-8). Moreover, the first genetic principal component became a significant factor (p = 7.65e-5) when analyzing frequent HMOs, while it had no effect on LMOs (p = 0.92) (Table 2).
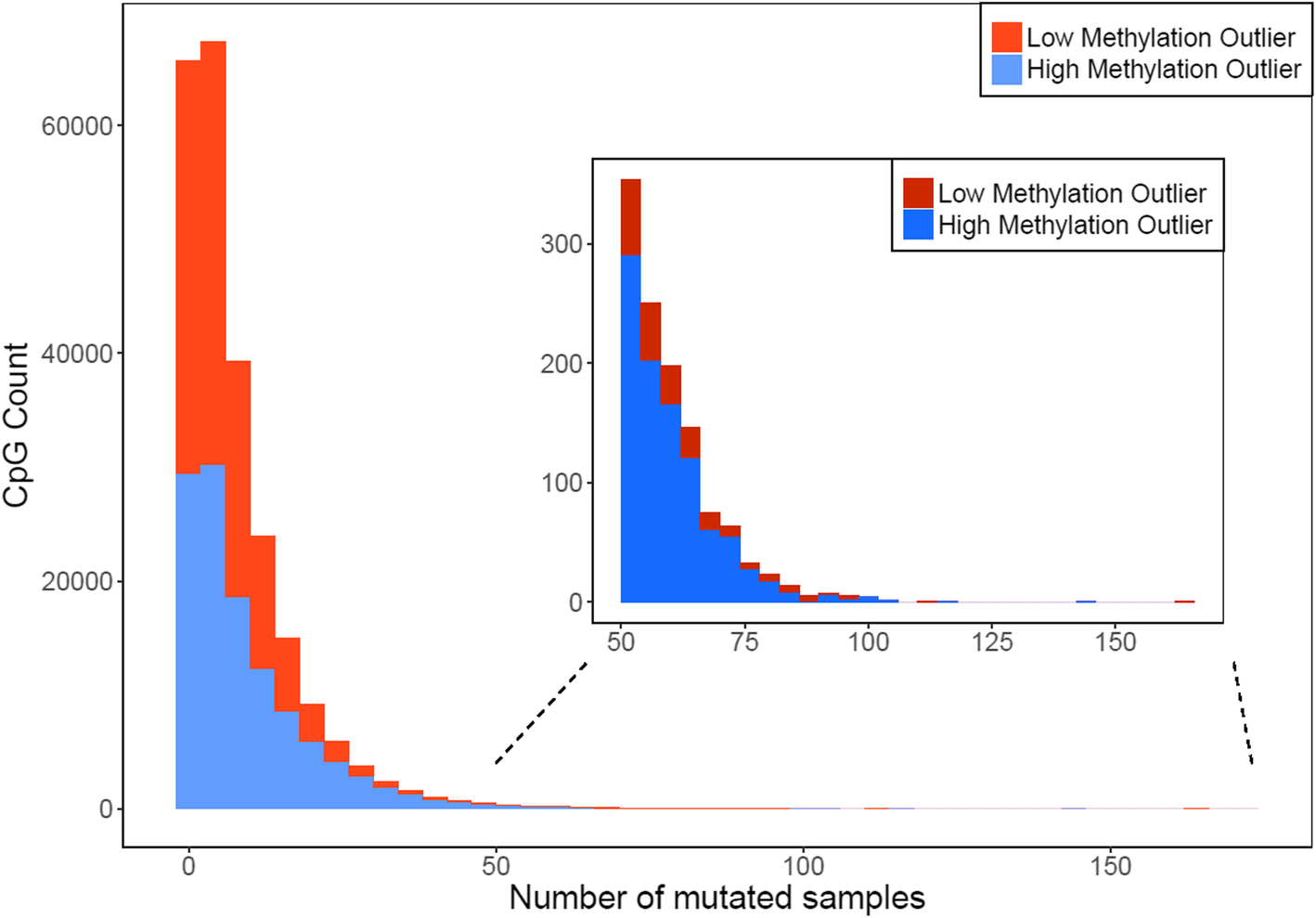
Fig. 2
The distribution of mutated samples for high methylation outliers (HMOs) and low methylation outliers (LMOs). For most CpGs, epigenetic mutations only occurred in a small number of samples, but HMOs were more likely to appear in a large number of samples (n > 50) than LMOs (969 HMOs and 216 LMOs, p < 1e-16).
To better present the longitudinal effect, the same analysis was performed on 110 individuals with four or more measures (in total 470 samples). Still, the total epigenetic mutations, frequent mutations, frequent HMOs, and LMOs all significantly increased with age (Fig. 1b, d, f) despite the lower statistical power (Additional file 1: Table S1). Among the other factors, sample quality and CD19 B cell proportion were still significantly associated with all the four outcomes, while the effects of sex and genetic PC1 were no longer significant (Additional file 1: Table S1).
Functional annotation of epigenetic mutations
To characterize HMO and LMOs, we examined their locations in relation to CpG island regions and regulatory features. Compared to all CpGs analyzed, where 33.5% of CpGs located in CpG islands, HMOs were enriched within CpG islands (63% of CpGs, p < 1e-16) and frequent HMOs even more so (88% of CpGs, p < 1e-16). On the other hand, LMOs were mostly located outside of CpG islands (88% CpGs outside of CpG islands, p < 1e-16), but the opposite was true for frequent LMOs, which were enriched in CpG islands (51% of CpGs, p = 8.6e-8) (Fig. 3). We further explored regulatory features of the frequent epigenetic mutations using the Ensembl database [14], and found that frequent HMOs were enriched in promoter regions (p = 1.1e-10), but less likely to be found in CCCTC-Binding factor (CTCF) binding sites (p = 1.4e-09) and regions of open chromatin (p = 3.6e-07) (Fig. 4a). The frequent LMOs, on the other hand, were enriched in CTCF (p = 7.7e-12) and transcription factor binding sites (p = 3.9e-05), open chromatin (p = 0.0012), and promoter flanking regions (p = 0.041), while depleted in promoter regions (p = 6.9e-19) (Fig. 4b). Moreover, we performed a pathway analysis of frequent epigenetic mutations using DAVID [15], but failed to identify enriched pathway.
.../...
F O R T H E R E S T O F T H E S T U D Y, P L E A S E V I S I T T H E S O U R C E .
.















