
































.
Thought leaders predict the tech developments that could have a big impact in the coming year.
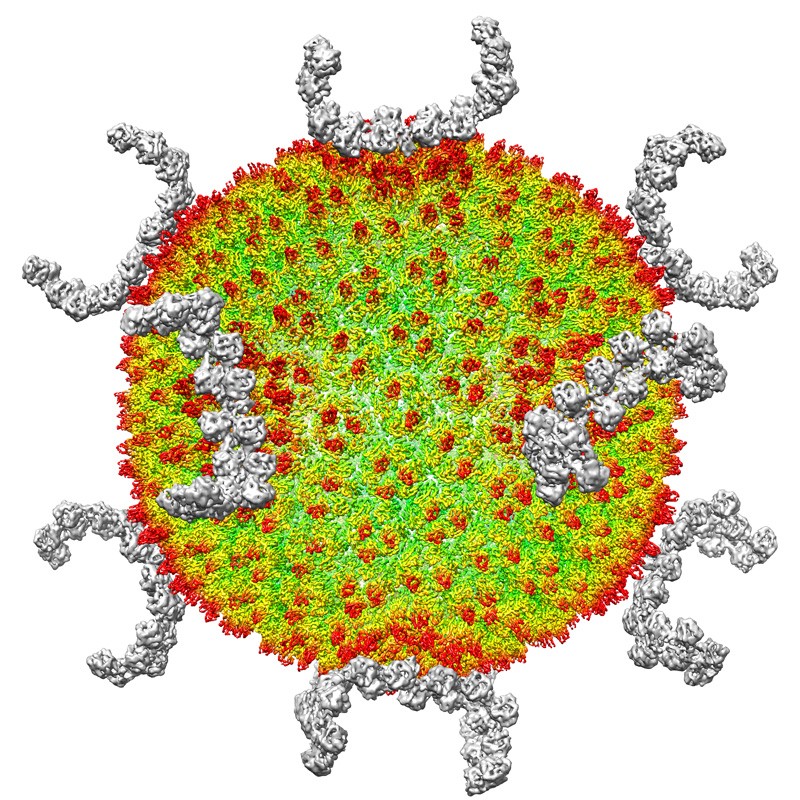
The virus SH1, reconstructed from images obtained using cryogenic electron microscopy.Credit: Luigi De Colibus/University of Oxford.
Better cryo-EM samples
Hongwei Wang Structural biologist at Tsinghua University in Beijing.
In two or three years, I think that transmission cryogenic electron microscopy (cryo-EM) will become the most powerful tool for deciphering the structures of macromolecules. These structures are crucial for understanding biochemical mechanisms and drug development, and methods for solving them more efficiently can speed up such work.
In cryo-EM, quickly freezing biological specimens in liquid nitrogen helps to preserve the molecules’ water content and reduces damage from the high-energy electrons used for imaging. But specimen preparation is a major bottleneck: if you don’t have a good specimen, you have nothing to image. Biological specimens often contain proteins, which unravel at the surface of the thin liquid layers used in the freezing process.
To prevent this unfolding, researchers are developing approaches that anchor proteins on to two-dimensional materials — such as the carbon lattice graphene — before applying the liquid droplets. That way, they can make the droplets even smaller while keeping the protein away from the air–water interface1.
Some laboratories place nanolitre-sized samples directly on to a surface2, instead of using cumbersome older methods that draw excess liquid away from larger droplets. Other methods use a focused ion beam to slice frozen cells into layers thinner than 100 nanometres, allowing researchers to study molecules in their cellular contexts3.
Solving a molecular structure with cryo-EM typically requires collecting and analysing as many as 10,000 images, representing several weeks to a month of work. Many images are imperfect, so we have to discard them. But theoretically, a few dozen pictures should be enough, and it would take less than a day to collect and analyse them. This increased throughput could help us to understand disease mechanisms and develop drugs more efficiently.
Improving RNA analysis
Sarah Woodson Biophysicist at Johns Hopkins University in Baltimore, Maryland.
I’m keeping my eye on long-read RNA sequencing and live-cell imaging using light-up RNA strands called aptamers. These technologies are still maturing, but I expect big changes in the next year or two.
Short-read sequencing has changed the field of RNA biology — it can tell you which RNA sequences contain a biochemically modified residue, for example. However, longer reads (for instance, using sequencing technologies offered by Oxford Nanopore and Pacific Biosciences) can now help to determine how common a particular modification is in the cell, and whether changes in one part of an RNA molecule correlate with changes in another.
Light-up aptamers are single-stranded DNA or RNA molecules that were developed in the lab to bind to fluorescent dyes. They are RNA analogues of the green fluorescent protein that is produced in some marine animals, and when these aptamers bind to the dyes, their fluorescence intensity increases. This enables researchers to track, for example, the formation of intracellular RNA clusters that contribute to neurodegenerative diseases.
Earlier light-up aptamers were unreliable: their signals were dim, and sometimes the aptamers didn’t work at all because the sequences misfolded when fused with the target RNA. But several groups have developed new types of fluorescent RNA, and in papers and talks I’ve seen a huge push to improve the brightness of existing aptamers and create variants that glow in different colours.
My lab has used chemical footprinting methods to study RNA folding in the cell. Many disorders are associated with changes in RNA structure, but that has been really hard to tease apart. Now we are turning to long-read sequencing and light-up aptamers to study RNA–protein aggregates in diseases including cancer, metabolic syndromes and Alzheimer’s. Using these technologies, we can better correlate cell death and other disease features with what’s happening to RNA molecules in the cell.
Decoding the microbiome
Elhanan Borenstein Computational systems biologist at Tel Aviv University, Israel.
Over the past decade, methods for sequencing the genetic content of microbial communities have probed the composition of the human microbiome. More recently, scientists have tried to learn what the microbiome is doing by integrating information about genes, transcripts, proteins and metabolites. Metabolites are especially interesting: they could offer the closest understanding of how the microbiome affects our health, because many host–microbiome interactions occur through the metabolites that bacteria generate and consume.
There has been an explosion of micro-biome–metabolome studies looking at, for instance, a set of stool samples — identifying the species present in each sample and their abundances through metagenomic sequencing, and using mass spectrometry and other technologies to measure the concentrations of different metabolites. By combining these two profiles, the hope is to understand which member of the microbiome is doing what, and thus whether specific microbes determine the level of certain metabolites.
But these data are complex and multi-dimensional, and there might be a whole web of interactions, involving multiple species and pathways, which ultimately produce a set of metabolites. Scientists have published computational methods to link microbiome and metabolome data and to learn these quirks and patterns. Such methods range from simple correlation-based analyses to complex machine-learning approaches that use existing microbiome–metabolome data sets to predict the metabolome in new microbial communities, or to recover microbe–metabolite relationships.
Our lab takes a different strategy. Rather than apply statistical methods to find microbe–metabolite associations, we build mechanistic models of how we think a specific microbial composition affects the metabolome, and use these as part of the analyses themselves. In effect, we are asking: on the basis of genomic and metabolic information, what do we know about each microbe’s ability to produce or take up specific metabolites? We can then predict the potential of a given collection of microbes to produce or degrade specific metabolites, and compare those predictions with actual metabolomic data. We showed that this approach avoids the pitfalls of simple correlation-based analyses4, and will release a new version of the analysis framework in the coming months.
Such studies could improve micro-biome-based therapies by identifying, for example, specific microbes responsible for producing too much of a harmful metabolite or too little of a beneficial one.

Software code can be used to build models that simulate tumour development.Credit: Getty.
Computing cancer
Christina Curtis Computational and systems biologist at Stanford University, California.
When it comes to cancer, we cannot see the process by which the disease forms, only its end point: we sample a tumour when it has become clinically detectable. By then, the tumour has acquired many mutations, and we’re left to work out what happened.
Our team built a computational model to explore the dynamics of tumour progression while accounting for tissue spatial structure. With this model, you can simulate a range of scenarios and generate ‘virtual tumours’ with patterns of mutation that mimic patient data. By comparing simulated data with actual genomic data, it’s possible to infer which parameters probably gave rise to a patient’s tumour.
I’m excited about complementing these inferential approaches with direct measurements of tumour lineage and phenotype using emerging barcoding and recording methods. Advances in the past two years include evolving CRISPR-based barcodes that can record the fate of cells during mammalian development5,6. Other techniques use image-based detection of DNA barcodes through in situ expression of RNA, thereby capturing cellular lineage, spatial proximity and phenotypes7.
In a study that modelled the growth of tumours in colon cancer8, we used tumour sequence data and simulations to study relationships between primary and metastatic tumours. These inferential analyses indicated that the vast majority of cancers had spread when the primary tumour comprised barely 100,000 cells — too small to detect using standard diagnostic methods such as colonoscopy.
With better sensitivity and scalability, a blend of modelling and measurement methods could track both lineage and spatial relationships during tumour formation, giving insight into cancer’s origins, including how specific mutations influence cellular fitness and fuel the disease’s progression.
Enhancing gene therapy
Alex Nord is a geneticist at the University of California, Davis.
We’re now about 15 years into large-scale experiments to map enhancers and other regulatory DNA sequences that control how genes are read out by cells and organs. Although more work is needed to complete these maps, we’re at the point at which we can harness our understanding to control the genome more precisely.
At the Society for Neuroscience annual meeting last October in Chicago, Illinois, I co-chaired a session that focused on identifying enhancer sequences and using them to control gene expression in specific cell types in the brain. One approach delivers engineered viruses into the brain to test thousands of enhancers for the gene-expression profile of interest. In 2019, researchers at the Allen Institute for Brain Science in Seattle, Washington, used this strategy to look for enhancers in specific cortical layers in the human brain9. And a team from Harvard University in Cambridge, Massachusetts, used an RNA-sequencing-based method to find enhancers that act only in specific interneurons, a type of nerve cell that creates circuits10.
Once enhancer sequences are identified, scientists can use them to drive expression in particular cell types for gene-therapy applications. In disorders caused by the inactivation or deletion of one copy of a gene, CRISPR–Cas9 gene-editing tools can target transcriptional activators to the gene’s enhancer to turn up expression of the working copy. Research in mice suggests these approaches can correct gene-expression deficiencies that lead to obesity and to conditions such as fragile-X, Rett and Dravet syndromes11 — the latter a severe form of epilepsy that my lab is working on. In the coming year, I think we’ll still be curing mice, but there is a lot of industry investment in this technology. The hope is that we can use these methods to transform how gene therapy is done in humans.
Single-cell sequencing
J. Christopher Love Chemical engineer at the Koch Institute for Integrative Cancer Research at MIT in Cambridge, Massachusetts.
I’m interested in how we bring medicines to patients faster and more accessibly. The technologies required are multifaceted. On the one hand, there’s discovery — for example, single-cell sequencing methods. On the other hand, there’s the matter of getting the technology to the patient — the manufacturing part. This is particularly relevant to medicines for rare diseases or for small populations, and is even applicable to global access to medicines we already have.
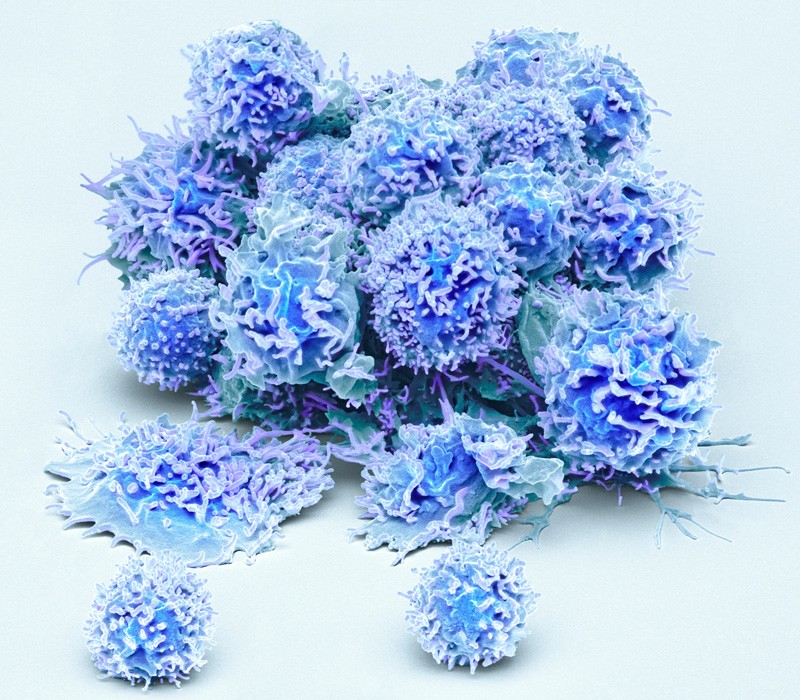
Activated T cells from human blood.Credit: Steve Gschmeissner/SPL.
On the discovery front, we’ve worked with colleagues at the Massachusetts Institute of Technology (MIT) in Cambridge to develop a portable, inexpensive platform for high-throughput, single-cell RNA sequencing12. But it’s still challenging to get sufficient resolution to distinguish between immune-cell subtypes, for instance, with different roles and antigen specificities. Over the past year or so, we’ve enhanced single-cell genomic sequencing in several ways. First, we came up with a method for detecting low-expression transcripts more efficiently13. And for T lymphocytes specifically, we designed a protocol that links each cell’s gene-expression profile with the sequence of its unique antigen receptor14.
Meanwhile, a team at the Dana Farber Cancer Institute in Boston, Massachusetts, has published a clever library-screening strategy to address the other side of the equation — working out which antigen a particular T-cell receptor recognizes15.
With MIT collaborator Alex Shalek and others, I have started a company, Honeycomb Biotechnologies, to commercialize our single-cell RNA-sequencing platform. Instead of having to spin down cells in a centrifuge, stick them in a tube, freeze it in liquid nitrogen and ship it from Africa, say, you could just ship an array of single-cell-sized wells — something the size of a USB thumb drive. That could make single-cell storage and genomic profiling possible for just about any sample anywhere in the world.
Linking genome structure and function
Jennifer Phillips-Cremins Epigeneticist and bioengineer at the University of Pennsylvania, Philadelphia
When you stretch out a single cell’s DNA end to end, it’s roughly 2 metres long — yet it has to fit into a nucleus with a diameter smaller than the head of a pin. The folding patterns cannot be random; chromosomes form 3D structures that must be spatially and temporally regulated across an organism’s lifespan.
.../...
.











