































.
F U L L T E X T S O U R C E : frontiers in Physiology
Aim:
Aging in humans is associated with a 10–40-fold greater incidence of sudden cardiac death from malignant tachyarrhythmia. We have reported that thiol oxidation of ryanodine receptors (RyR2s) by mitochondria-derived reactive oxygen species (mito-ROS) contributes to defective Ca2+ homeostasis in cardiomyocytes (CMs) from aging rabbit hearts. However, mechanisms responsible for the increase in mito-ROS in the aging heart remain poorly understood. Here we test the hypothesis that age-associated decrease in autophagy is a major contributor to enhanced mito-ROS production and thereby pro-arrhythmic disturbances in Ca2+ homeostasis.
Methods and Results:
Ventricular tissues from aged rabbits displayed significant downregulation of proteins involved in mitochondrial autophagy compared with tissues from young controls. Blocking autophagy with chloroquine increased total ROS production in primary rabbit CMs and mito-ROS production in HL-1 CMs. Furthermore, chloroquine treatment of HL-1 cells depolarized mitochondrial membrane potential (Δψm) to 50% that of controls. Blocking autophagy significantly increased oxidation of RyR2, resulting in enhanced propensity to pro-arrhythmic spontaneous Ca2+ release under β-adrenergic stimulation. Aberrant Ca2+ release was abolished by treatment with the mito-ROS scavenger mito-TEMPO. Importantly, the autophagy enhancer Torin1 and ATG7 overexpression reduced the rate of mito-ROS production and restored both Δψm and defective Ca2+ handling in CMs derived from aged rabbit hearts.
Conclusion:
Decreased autophagy is a major cause of increased mito-ROS production in the aging heart. Our data suggest that promoting autophagy may reduce pathologic mito-ROS during normal aging and reduce pro-arrhythmic spontaneous Ca2+ release via oxidized RyR2s.
Introduction
Sudden cardiac death due to malignant ventricular tachyarrhythmia remains a major cause of mortality in the United States, with ∼300,000 cases each year (Deo and Albert, 2012). Risk of sudden cardiac death increases 5–40-fold in people over 65 years old compared to younger individuals (Zheng et al., 2001; Podrid and Myerburg, 2005). We used the aged rabbit model to study the mechanisms of arrhythmogenesis in aging humans. The rabbit heart closely resembles the human heart in its electrical and contractile properties (Cooper et al., 2012). Our studies of aged rabbit hearts (>4 years old) revealed conduction abnormalities, myocardial stiffening, increased interstitial fibrosis, slowed transverse conduction, and depletion of the Purkinje network (Cooper et al., 2012). Moreover, we demonstrated that aged rabbit cardiomyocytes (CMs) have depolarized mitochondrial membrane potentials (Δψm), higher levels of mitochondria-derived reactive oxygen species (mito-ROS), increased calcium (Ca2+) leak from the mito-ROS-oxidized type 2 ryanodine receptor (RyR2), and increased formation of pro-arrhythmic spontaneous Ca2+ waves (SCWs) (Cooper et al., 2013). Mitochondria participate in a variety of intracellular tasks including energy production and nutrient sensing (Dai et al., 2012). Excessive production of ROS by defective mitochondria is proarrhythmogenic and predisposes the aging heart to lethal ventricular tachyarrhythmia (Zorov et al., 2000; Janczewski and Lakatta, 2010; Cooper et al., 2013; Hamilton and Terentyev, 2019).
Intracellular Ca2+ release from the SR mediated by RyR2 is a critical determinant of cardiac contractility. Abnormally high RyR2 activity has been implicated in arrhythmogenesis and is present in a variety of diseases such as heart failure and myocardial infarction (Wehrens et al., 2005; Belevych et al., 2011, 2012; Plummer et al., 2011; Zima et al., 2014). We have demonstrated that hyperactivity of RyR2 in aged hearts is caused by oxidation of reactive cysteines in the channel by excessive ROS emitted by mitochondria and that treatment of aged myocytes with the mitochondria-specific scavenger mito-TEMPO effectively restores intracellular Ca2+ cycling (Cooper et al., 2013). However, the mechanisms underlying enhanced mito-ROS generation and concomitant pro-arrhythmic changes in Ca2+ cycling, characteristic of aging, are not well understood.
The mitochondrial population is a vast network controlled by cycles of fission and fusion and is regulated by mitophagy or autophagy at the organelle level. Fission is a culling mechanism for damaged mitochondria, while fusion recombines fragmented, healthy mitochondria into larger, bio-energetically favorable structures. Autophagy sequesters unnecessary or dysfunctional cellular components (including proteins, organelles, and even bacteria) in a double-membrane structure called an autophagosome that is then degraded by lysosomal acidic hydrolases (Ravikumar et al., 2009; Ashrafi and Schwarz, 2013). Damaged smaller mitochondria produced by fission are cleared from the mitochondrial network by mitophagy, which is a self-degradative catabolic process (Kubli and Gustafsson, 2012; Ashrafi and Schwarz, 2013). Mitophagy involves upregulation of general autophagy machinery components followed by the recognition of damaged mitochondria as targets for clearance. Without efficient autophagy (and its mitochondria-specific form, mitophagy), damaged mitochondria would remain in the cell and generate harmful ROS (Miquel et al., 1980; Terman et al., 2010; Sharp et al., 2014). Because both autophagy and mitophagy are impaired in the aged heart (Taneike et al., 2010; Dai et al., 2012; Lopez-Otin et al., 2013), we hypothesized that decreased autophagy may predispose the aging heart to arrhythmogenesis.
Here we show that inhibition of autophagy with chloroquine, which blocks lysosomal acidification and prevents autophagosome fusion and therefore subsequent degradation (Shintani and Klionsky, 2004), promoted generation of SCWs at the cellular scale because of RyR2 oxidation. By contrast, enhancing autophagy in cultured primary aged myocytes using Torin1, a potent inhibitor of the negative autophagy regulator mTOR (Thoreen et al., 2009), reduced the frequency of spontaneous Ca2+ release. In addition to pharmacologic experiments, to enhance autophagy we overexpressed ATG7, an essential component of the autophagosome that is sufficient to increase autophagy flux (Bhuiyan et al., 2013; Martinez-Lopez et al., 2015). The results of these overexpression experiments showed a significant reduction in age-related instances of spontaneous Ca2+ release. We conclude that defective autophagy contributes to the age-associated increase in mito-ROS production and pro-arrhythmic Ca2+ mishandling.
Results
Autophagy Is Downregulated in the Aging Rabbit Heart
First we examined the ultrastructure of CMs isolated from both young and aged left ventricular free wall (Cooper et al., 2012, 2013; Morrissey et al., 2017) using transmission electron microscopy. Electron microscopy imaging show that the arrangement of mitochondria in aged CMs is both disorganized and fragmented (Figure 1A). Mitochondrial size analysis was performed using the GLIMMIX procedure in SAS (SAS, Inc.) of 71 sections from four aged and four young hearts (up to 13 sections per heart). Comparing the cumulative distributions of mitochondrial populations revealed a narrower (leptokurtic) curve for young mitochondria and a wider (platykurtic) curve for aged mitochondria (Figure 1B). Further, aged CMs contain a mitochondrial population with increased variance in cross-sectional area compared to young CMs (0.6 vs. 0.9 μm2) (p < 0.05) (Figure 1C). These observations indicate that mitochondria from aged CMs vary in size to a greater degree than mitochondria from young CMs. We then performed western blot analysis against proteins important in mitochondrial regulation of fission (DRP1) and fusion (MFN2) to better understand why mitochondria in aged CMs are more heterogeneous. Immunoblots with homogenates prepared from left ventricular free walls revealed 2.25-fold higher expression of DRP1 in aged hearts relative to young samples (p < 0.05, Figure 1D), but no difference in expression of MFN2. We then performed western blot analysis of homogenates prepared from young and aged hearts against markers of autophagy and mitophagy to understand why defective mitochondria remain in aged CM. The level of the autophagosome scaffolding protein p62 was 1.5-fold higher in aged samples (p < 0.05) (Ohsumi, 2001; Hoshino et al., 2013). In addition, the light-chain 3 cleavage ratio (II/I) was 1.7-fold higher in aged compared to young samples (p < 0.01), consistent with decreased autophagy. Levels of PINK1, the ubiquitin-mediated mitophagy kinase, were 20% lower in aged relative to young samples (p < 0.05). The transcriptional autophagy effector p53 was 2.5-fold more abundant in aged compared to young samples (p < 0.05) (Figure 1E). Next, to mimic aging-related reduction in autophagy we administered the autophagy inhibitor chloroquine to young primary rabbit myocytes (600 nM, 3 h). Using the ROS-specific fluorescent indicator DCFDA, we found markedly higher ROS levels as well as ROS production rate (0.19 vs. 0.38 F∗s–1, p < 0.05) (Figure 1F). Overall, we also found that mitochondria in aged CMs have greater variance in mitochondrial cross-sectional area and display protein profiles consistent with decreased autophagy and increased fission. Further, pharmacologically blocking autophagy in CMs from young animals dramatically increased ROS, making the young CMs more closely resemble aged CMs.
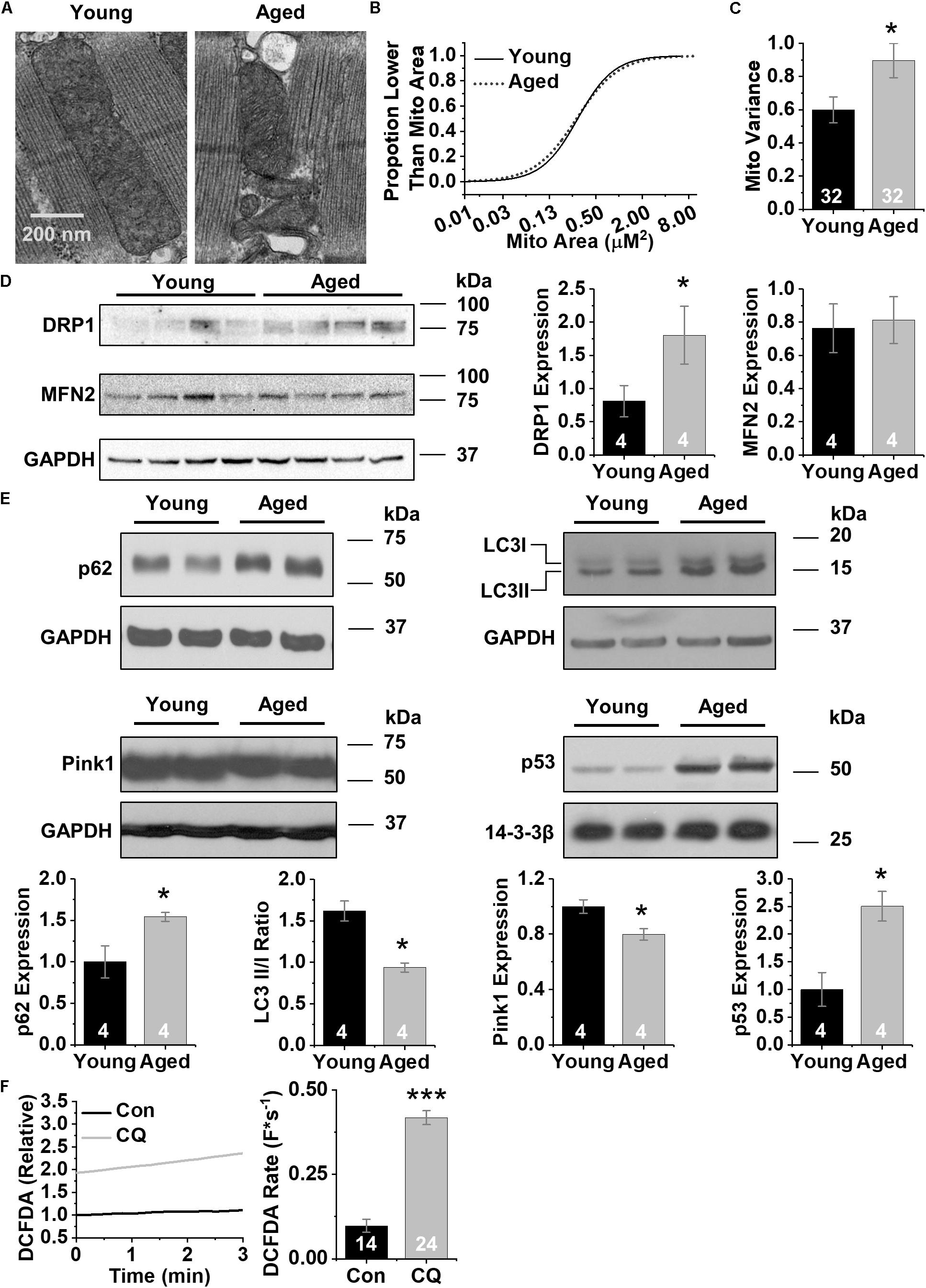
Figure 1. CM aging leads to heterogeneous mitochondria and decreased markers of autophagy. (A) Representative transmission electron micrographs of acutely disassociated CMs from young and aged rabbit hearts, scale bar = 200 nm. (B) Cumulative distribution of mitochondrial log2 cross-sectional areas assessed by image analysis of electron micrographs in young and aged CMs. Analysis of data was performed using the GLIMMIX procedure in SAS Version 9.4 (SAS Inc.) (∗p < 0.05). © Variance in mitochondrial cross-sectional areas. (D) Representative immunoblots for mitochondrial fission and fusion proteins, DRP1 and MFN2 with densitometry using young and aged whole left ventricular homogenates. (E) Representative immunoblots for autophagy (p62, LC3) and mitophagy proteins (Pink1 and p53) using young and aged whole left ventricular homogenates. (F) Changes in DCFDA fluorescence in chloroquine- and vehicle-treated CMs. Mean data represent ROS production rate. ∗p < 0.05, and ∗∗∗p < 0.001 all values are mean ± SEM.
Autophagy Inhibition Decreases Δψm, Increases Mito-ROS, and Promotes Spontaneous Ca2+ Release in HL-1 CMs
To dissect out the effects of decreased autophagy during cardiac aging on dysfunctional Ca2+ handling, we treated cells from the established HL-1 atrial CM cell line (Claycomb et al., 1998) with 600 nM chloroquine or vehicle control for 3 h. At baseline HL-1 CMs produced low levels of mito-ROS. Compared with control cells, chloroquine-treated cells exhibited lower autophagy flux (shown as a high ratio of positive puncta in Figure 2A) using adenovirus containing TF-LC3-GFP. The ratio of red fluorescence (autolysosomes) to green fluorescence (autophagosomes) decreased 44% after the addition of chloroquine, indicating less autophagy. Western blot analysis reveals that these cells expressed 1.75-fold more p62 than did control cells (p < 0.05) (Figure 2B). General ROS was measured using the fluorescent indicator CM-H2DCFDA. Chloroquine treatment increased ROS generation compared to controls (0.23 ± 0.06 vs. 0.13 ± 0.09 F∗s–1) (p < 0.05), and ROS generation was rescued by the application of the mito-ROS scavenger mito-TEMPO (0.16 ± 0.07 F∗s–1) (p < 0.05) (Liu et al., 2010; Figure 2C). Direct measurement of mito-ROS using the specific fluorescent superoxide indicator MitoSOX showed that chloroquine treatment induced a 43% increase in mito-ROS (100 ± 20 vs. 143 ± 50%) (Figure 2D, p < 0.05). To confirm our results with fluorescent indicator dyes, we used the ratiometric mitochondrial H2O2 biosensor MTroGFP-ORP1 to specifically measure the level of fractional oxidation due to mitochondrial H2O2 as previously described (Dey et al., 2018). Mitochondrial H2O2 levels increased after chloroquine treatment compared to vehicle control (0.26 ± 0.01 vs. 0.30 ± 0.01%, p < 0.05) (Figure 2E). Live cell measurements of Δψm using the fluorescence indicator TMRM revealed a chloroquine-mediated decrease in Δψm compared to vehicle control (2.1 ± 0.4 vs. 1.58 ± 0.6 [Math Processing Error]) (p < 0.05) (Figure 2F). These data indicate that autophagy blockade in HL-1 CMs decreases Δψm and increases mito-ROS.
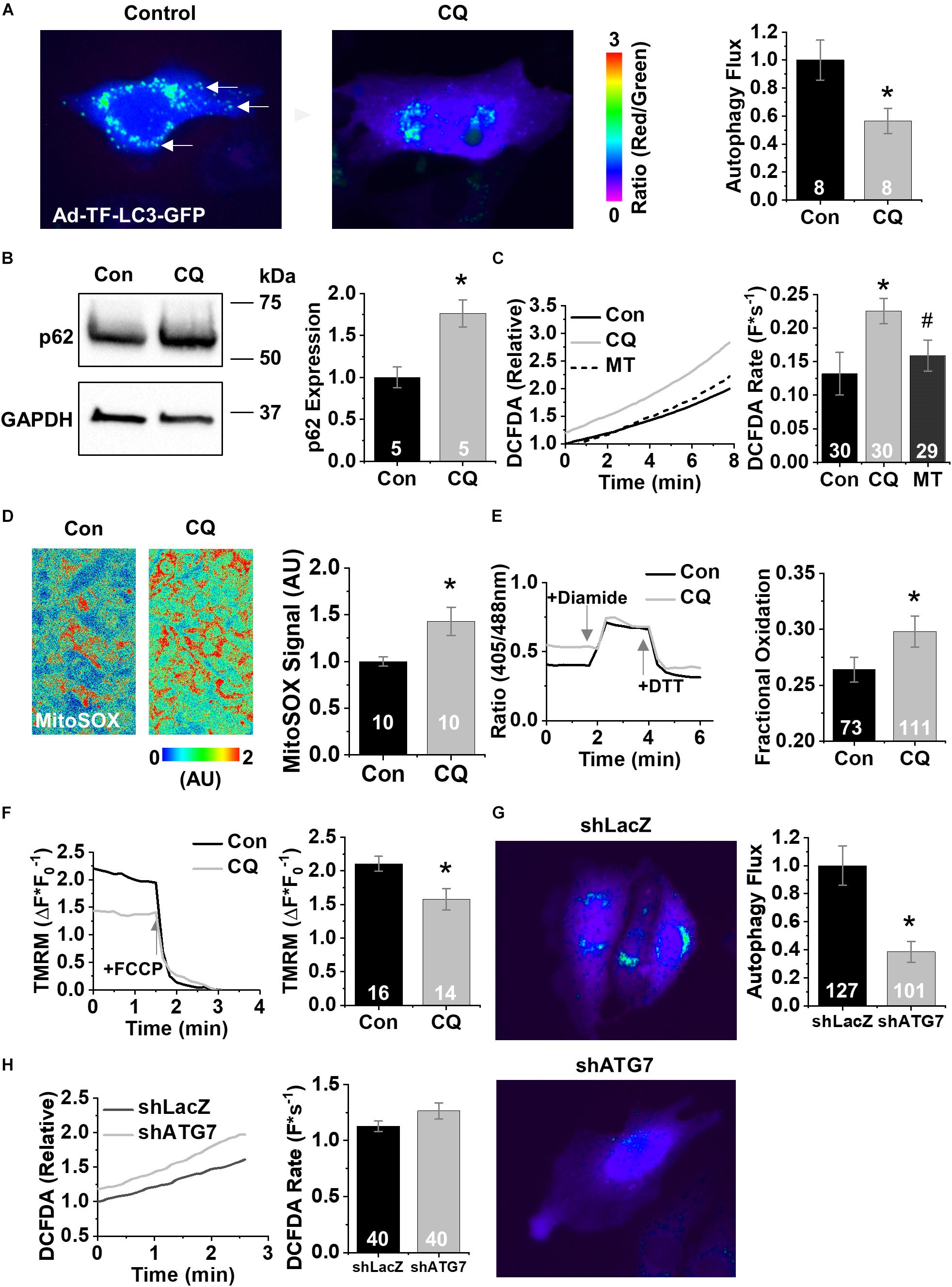
Figure 2. Chloroquine decreases autophagy in HL-1 CMs. (A) Fluorescence images of HL-1 CMs infected with Ad-TF-LC3-GFP visualizing autolysosomes as puncta. Examples of autophagosomes are shown by white arrow. (B) Representative immunoblots for the autophagy protein p62. © Changes in DCFDA fluorescence in chloroquine- and vehicle-treated CMs, and from CMs treated with 25 μM mito-TEMPO for 10 min following chloroquine treatment. Mean data represent ROS production rate. (D) Representative confocal images of HL1 cells stained with MitoSOX acquired in XYT mode. Relative MitoSOX fluorescence is plotted. (E) Representative line traces from confocal imaging of MTroGFP-ORP1. Mean data represent fractional oxidation derived as baseline normalized to fluorescence minimum in the presence of DTT and fluorescence maximum in the presence of diamide. (F) Traces and mean data of normalized TMRE fluorescence of chloroquine- and vehicle-treated CMs. Data are normalized to the minimum TMRM fluorescence in the presence of the uncoupling agent FCCP. (G) Mean data represent TF-LC3-GFP flux data after treatment with short hairpins against LacZ or ATG7. (H) Changes in DCFDA fluorescence after short hairpin treatment. Mean data represent ROS production rate. ∗p < 0.05, all values are mean ± SEM.
To confirm these pharmacological findings, we next tested the effect of genetic ablation of autophagy using short hairpin RNAi (shRNA) against the essential autophagy enzyme ATG7 (shATG7) (Bhuiyan et al., 2013). Treatment of HL-1 CMs with shATG7 resulted in lower autophagy flux compared to that of LacZ control shRNA (1.0 ± 0.14 vs. 0.39 ± 0.07, p < 0.05), measured using adenovirus containing TF-LC3-GFP (Figure 2G). HL-1 CMs tolerated the shATG7 treatment, and analysis of ROS production after RNAi showed a strong trend toward ATG7 increasing ROS production (1.13 ± 0.05 vs. 1.26 ± 0.07 F∗s–1, p = 0.058) (Figure 2H). To test the functional effects of decreased autophagy on Ca2+ handling, we performed live-cell Ca2+ imaging of HL-1 CMs with 1 Hz field-stimulation (Figure 3A). Quantification of Ca2+ handling parameters revealed that the proportion of cells exhibiting pro-arrhythmic global SCWs was markedly increased after autophagy block with chloroquine (58% of control cells exhibited SCWs vs. 95% of chloroquine-treated cells, p < 0.05) (Figure 3B). Further, the latency at which SCWs formed was significantly shorter with chloroquine treatment compared to control (1.9 ± 1.1 vs. 8.8 ± 5.8 s) (p < 0.05). Because multiple physiological changes may be elicited by blocking autophagy with chloroquine, we applied the superoxide scavenger mito-TEMPO (25 μM, 10 min) (Ni et al., 2016) to HL-1 CMs treated with chloroquine. Though there was no change in the proportion of cells exhibiting SCWs, scavenging mito-ROS significantly improved the latency to SCW compared to chloroquine (5.6 ± 3.2 vs. 1.9 ± 1.1 s) (p < 0.05).
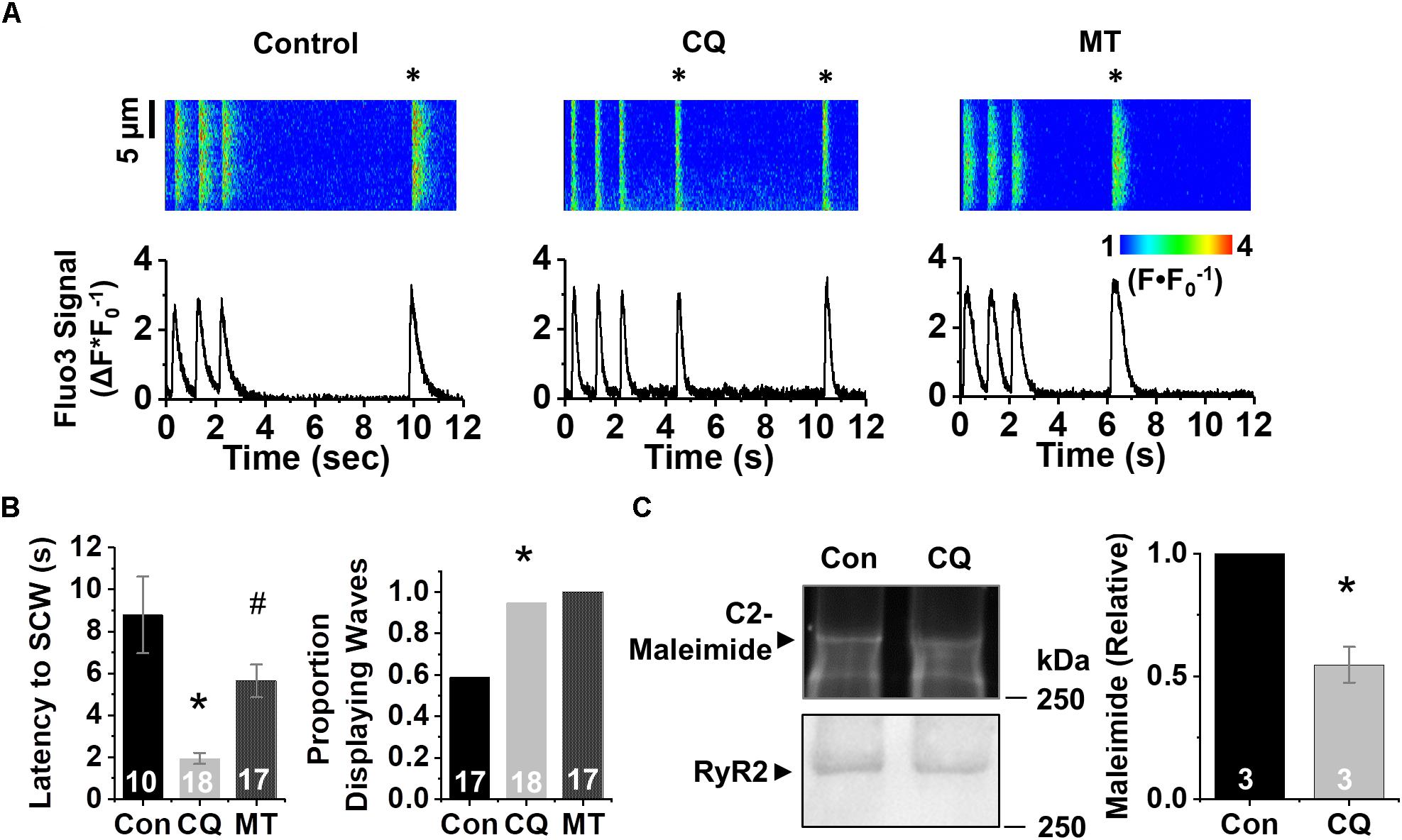
Figure 3. Inhibition of autophagy reduces refractoriness of RyR2-mediated Ca2+ release under β-adrenergic stimulation. (A) Confocal line scan images and Fluo3 profiles at the end of 1 min 1 Hz pacing train with HL-1 CMs treated with chloroquine or vehicle; all with 100 nM isoproterenol. SCWs are indicated by an asterisk. (B) Mean data for SCW latency and proportion of cells exhibiting SCWs (proportion data ∗p < 0.05, Fisher’s exact test). © Representative C2-maleimide fluorescence of RyRs from HL-1 CMs treated with chloroquine or vehicle. Relative maleimide fluorescence scored between conditions. n = 3 biological preparations from sequential passages. ∗p < 0.05, all values are mean ± SEM. #p < 0.05 compared to control.
Given the increase in SCW formation in HL-1 CMs after autophagy block, we assessed the levels of oxidation of the RyR2. Protein lysates conjugated with C2-maleimide (Figure 3C), a free thiol fluorescence indicator (Hanna et al., 2014), were resolved in non-reducing conditions using PAGE. After C2-maleimide fluorescence signal was imaged, immunoblots were performed on the same gel to obtain RyR2 signal, which was in turn used to normalize the C2-maleimide signal against total RyR2. Densitometry showed a twofold reduction in maleimide signal, indicating that blocking autophagy increases thiol oxidation on the RyR2 (p < 0.05).
.../...
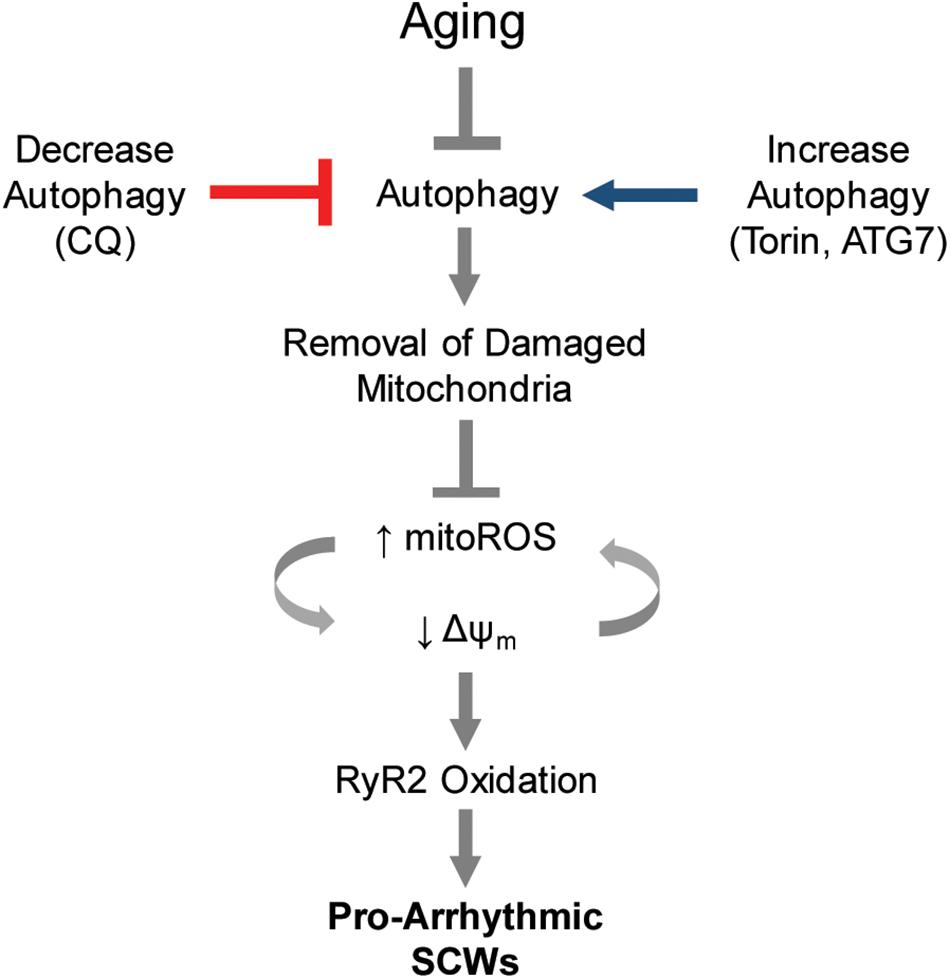
Figure 6. Proposed model of autophagy control of Ca2+-mediated cardiac arrhythmia. Autophagy is a major cause of increased mito-ROS production in the aging heart. Blocking or enhancing autophagy is sufficient to modify pathologic mito-ROS production and Δψm during normal aging, thereby controlling the instance of pro-arrhythmic spontaneous Ca2+ release via oxidized RyR2s.
.../...
.














