































.
F U L L T E X T S O U R C E : PLOS_GENETICS
THIS IS AN UNCORRECTED PROOF!
Abstract
'Epigenetic age acceleration' is a valuable biomarker of ageing, predictive of morbidity and mortality, but for which the underlying biological mechanisms are not well established. Two commonly used measures, derived from DNA methylation, are Horvath-based (Horvath-EAA) and Hannum-based (Hannum-EAA) epigenetic age acceleration. We conducted genome-wide association studies of Horvath-EAA and Hannum-EAA in 13,493 unrelated individuals of European ancestry, to elucidate genetic determinants of differential epigenetic ageing. We identified ten independent SNPs associated with Horvath-EAA, five of which are novel. We also report 21 Horvath-EAA-associated genes including several involved in metabolism (NHLRC, TPMT) and immune system pathways (TRIM59, EDARADD). GWAS of Hannum-EAA identified one associated variant (rs1005277), and implicated 12 genes including several involved in innate immune system pathways (UBE2D3, MANBA, TRIM46), with metabolic functions (UBE2D3, MANBA), or linked to lifespan regulation (CISD2). Both measures had nominal inverse genetic correlations with father’s age at death, a rough proxy for lifespan. Nominally significant genetic correlations between Hannum-EAA and lifestyle factors including smoking behaviours and education support the hypothesis that Hannum-based epigenetic ageing is sensitive to variations in environment, whereas Horvath-EAA is a more stable cellular ageing process. We identified novel SNPs and genes associated with epigenetic age acceleration, and highlighted differences in the genetic architecture of Horvath-based and Hannum-based epigenetic ageing measures. Understanding the biological mechanisms underlying individual differences in the rate of epigenetic ageing could help explain different trajectories of age-related decline.
Author summary
DNA methylation, an epigenetic process, is known to vary with age. Methylation levels at specific sites across the genome can be combined to form estimates of age known as ‘epigenetic age’. The difference between epigenetic age and chronological age is referred to as ‘epigenetic age acceleration’, with positive values indicating that a person is biologically older than their years. Understanding why some people seem to age faster than others could shed light on the biological processes behind age-related decline; however, the mechanisms underlying differential rates of epigenetic ageing are largely unknown. Here, we investigate genetic determinants of two commonly used epigenetic age acceleration measures, based on the Horvath and Hannum epigenetic clocks. We report novel genetic variants and genes associated with epigenetic age acceleration, and highlight differences in the genetic factors influencing these two measures. We identify ten genetic variants and 21 genes associated with Horvath-based epigenetic age acceleration, and one variant and 12 genes associated with the Hannum-based measure. There were no genome-wide significant variants or genes in common between the Horvath-based and Hannum-based measures, supporting the hypothesis that they represent different aspects of ageing. Our results suggest a partial genetic basis underlying some previously reported phenotypic associations.
Introduction
Ageing is associated with a decline in physical and cognitive health, and is the main risk factor for many debilitating and life-threatening conditions including cardiovascular disease, cancer, and neurodegeneration [1]. Ageing is a multi-dimensional construct, incorporating physical, psychosocial, and biological changes. Everyone experiences the same rate of chronological ageing, but the rate of ‘biological ageing’, age-related decline in physiological functions and tissues, differs between individuals. Various phenotypic and molecular biomarkers have been used to study biological ageing, including a number of 'biological clocks', the best known of which is telomere length. Telomeres shorten with increasing age, and telomere length has been found to predict morbidity and mortality [2]. More recently, research into epigenetics–chemical modifications to DNA without altering the genetic sequence–has yielded another method for measuring biological age.
DNA methylation is an epigenetic modification, typically characterised by the addition of a methyl group to a cytosine-guanine dinucleotide (CpG) [3], that can influence gene expression and is associated with variation in complex phenotypes. This process is essential for normal development and is associated with a number of key processes including ageing. DNA methylation levels are dynamic, varying with age across the life course [4,5] and are influenced by both genetic and environmental factors [6].
Weighted averages of methylation at multiple CpG sites can be integrated into estimates of chronological age referred to as ‘epigenetic age’. Two influential studies have used this method to create ‘epigenetic clocks’, which accurately predict chronological age in humans. Hannum et al. used DNA methylation profiles from whole blood from two cohorts to identify 71 CpG sites that could be used to generate an estimate of age [7], while Horvath used data from 51 different tissue types from multiple studies to identify 353 CpG sites whose methylation levels can be combined to form an age predictor [8]. Hannum et al.’s clock is specific to blood samples, although it can be adjusted for different tissue types using linear models. The Horvath clock is widely applicable, with the same CpG set and the same algorithm being used irrespective of the DNA source.
Although similar penalised regression models were used to select the CpG sites to be included in each of these epigenetic clocks, there is limited overlap in the CpGs included. The two measures are clearly related, but are thought to capture slightly different aspects of the biology of ageing [9]. The Hannum age estimator correlates with proportions of certain blood cells, reflecting its construction based on blood methylation data [9,10], and it is considered to track aspects of immunosenescence. The pan-tissue Horvath clock, constructed across a broad spectrum of tissue and cell types, is relatively uncorrelated with blood cell proportions [11], and is thought to capture cell-intrinsic changes in DNA methylation which might reflect an innate ageing process.
Both the Hannum and Horvath epigenetic clocks are strongly correlated (r>0.95) with chronological age [7,8]. However, despite these high overall correlations, there can be substantial differences between epigenetic and chronological age at the individual level, and it is unclear what drives these differences. A greater epigenetic age relative to chronological age is commonly described as ‘epigenetic age acceleration’ (EAA), and implies that a person is biologically older than their years. EAA has been shown to be informative for both current and future health trajectories [9]. Recently, a growing number of studies have used EAA to investigate age-related disorders, and the epigenetic clock is increasingly being recognised as a valuable marker of biological ageing [10,12].
The simplest definition of epigenetic age acceleration is the residual that results from regressing epigenetic age on chronological age. However, it is well known that the abundance of different cell types in the blood changes with age [13,14], and hence two broad categories of EAA measures have been distinguished: those that are independent of age-related changes in blood cell composition, and those that incorporate and are enhanced by blood cell count information [10]. The former group, considered to reflect ‘pure’ epigenetic ageing effects that are not influenced by differences in blood cell counts, are often referred to as ‘intrinsic’ epigenetic age measures. The latter group up-weights the contributions of blood cell counts, thus leveraging known age-related changes to blood cell proportions to capture aspects of immunosenescence; these measures are referred to as ‘extrinsic’ epigenetic age measures.
In keeping with previous work, this study focuses on two different epigenetic age measures, based on the Horvath and Hannum epigenetic clocks [7,8], and uses these to derive variations of EAA that are either independent of blood cell counts, or enhanced by changes in blood cell composition. Horvath-based epigenetic age follows the approach by Horvath (2013), and is defined as the predicted value of age based on the DNA methylation levels of the 353 CpG sites identified in his study [8]. Horvath-based epigenetic age acceleration (Horvath-EAA) is the residual term of a multivariate model regressing the Horvath-based epigenetic age estimate on chronological age and estimates of blood cell counts. It is by definition independent of both chronological age and age-related changes in the cellular composition of blood. Hannum-based epigenetic age is based on DNA methylation levels at the 71 CpGs identified by Hannum et al. (2013) [7]. Hannum-based epigenetic age acceleration (Hannum-EAA) is an enhanced version of the Hannum estimate which up-weights the contributions of age-associated blood cells. A weighted average of Hannum-based epigenetic age with blood cells whose abundance is known to change with age is calculated, and Hannum-EAA is then defined to be the residual variation from a univariate model regressing the weighted DNA methylation age estimate on chronological age. Hannum-EAA is independent of chronological age but in addition to cell-intrinsic epigenetic changes it also tracks age-related changes in blood cells. Full details of the calculation of Horvath-EAA and Hannum-EAA are given in S1 Text.
Horvath-EAA, described in previous publications as ‘intrinsic’ epigenetic age acceleration (IEAA), can be interpreted as a measure of cell-intrinsic ageing that exhibits preservation across multiple tissues, appears unrelated to lifestyle factors, and probably indicates a fundamental cell ageing process that is largely conserved across cell types [8,10]. In contrast, Hannum-EAA, referred to in previous studies as ‘extrinsic’ epigenetic age acceleration (EEAA), can be considered a biomarker of immune system ageing, explicitly incorporating aspects of immune system decline such as age-related changes in blood cell counts, correlating with lifestyle and health-span related characteristics, and thus yielding a stronger predictor of all-cause mortality [10,15].
It should be noted that as both the Horvath and Hannum epigenetic clocks correlate well with age, in a population with a wide age range they are guaranteed to correlate with each other. However, Horvath-based and Hannum-based epigenetic age acceleration estimates, i.e. the degree of divergence of epigenetic age from chronological age, are not guaranteed to be correlated.
Previous studies have identified relationships between epigenetic ageing and numerous traits, including several age-related health outcomes, for example Alzheimer’s disease pathology [16], cognitive impairment [16], and age at menopause [17]. Higher EAA has been associated with poorer measures of physical and cognitive fitness [9] and higher risk of all-cause mortality [12]. Many associations are specific to either Horvath-EAA or Hannum-EAA, a discordance that may reflect the differences in the two estimates and supports the theory that they represent different aspects of ageing [15,18,19].
While EAA has been associated with various markers of physical and mental fitness, the mechanisms underlying epigenetic ageing remain largely unknown. There has been little research conducted thus far on genetic contributions to epigenetic age acceleration. However, Lu et al. (2018) recently published results of the first genome-wide association analysis of blood EAA in a sample of 9,907 individuals, identifying five genetic loci associated with Horvath-EAA and three Hannum-EAA-associated loci [20].
This current study, with a sample size of 13,493 individuals, constitutes the largest study of the genetic determinants of DNA methylation-based ageing to date. Single nucleotide polymorphism (SNP)-based and gene-based approaches were used to identify genes and loci associated with Hannum-based and Horvath-based estimates of EAA. Functional mapping and annotation of genetic associations were performed, alongside gene-based and gene-set analyses, in an attempt to elucidate the genes and pathways implicated in differential rates of epigenetic ageing between individuals and shed light on the underlying biological mechanisms. We report novel SNPs and genes associated with epigenetic age acceleration, and highlight differences in the genetic architectures of the Horvath-based and Hannum-based EAA measures.
Results
Estimation of epigenetic age and epigenetic age acceleration in the Generation Scotland sample
A summary of the estimated epigenetic age variables in Generation Scotland (GS) is given in S1 Table. Both the Horvath- and Hannum-based estimates of biological age were highly correlated with chronological age (r = 0.94, SE = 0.005 and r = 0.93, SE = 0.005 respectively). The two DNA methylation age estimates were also highly correlated with each other (r = 0.93, SE = 0.005); however, the two estimates of epigenetic age acceleration, Horvath-EAA and Hannum-EAA, were only weakly correlated (r = 0.30, SE = 0.013) (Fig 1).
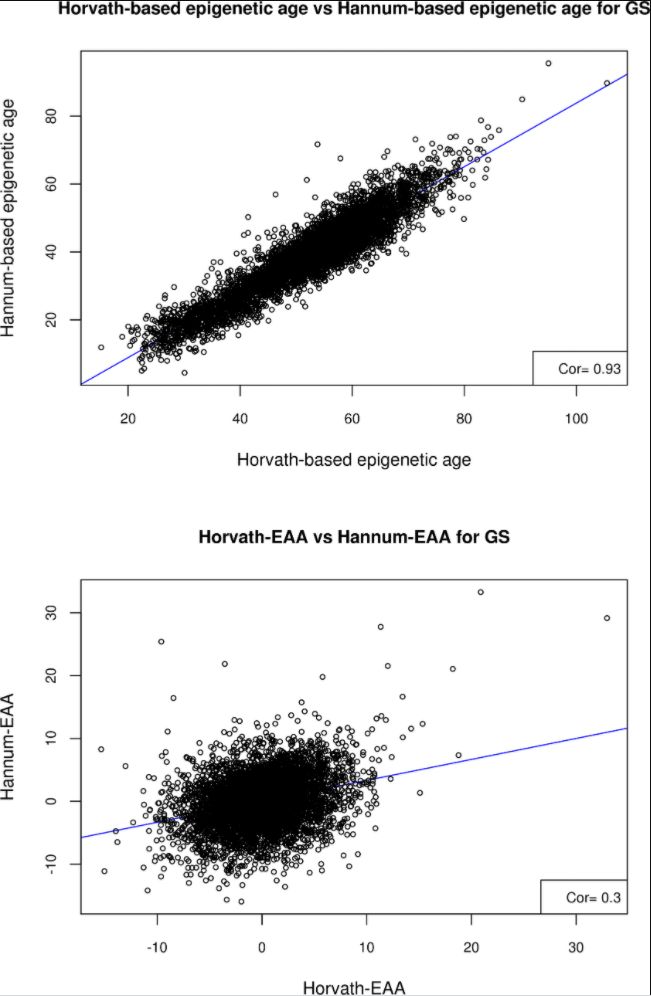
Fig 1.
Scatter plots of A) Horvath-based epigenetic age versus Hannum-based epigenetic age, and B) Horvath-EAA vs Hannum-EAA, for the Generation Scotland sample.
https://doi.org/10.1...en.1008104.g001
GWAS of Horvath-EAA and Hannum-EAA in GS and replication of previously identified loci
The genome-wide association study (GWAS) for the GS cohort yielded two significant (P<5x10-8) variants for Horvath-EAA, but no SNPs achieved genome-wide significance for association with Hannum-EAA (minimum P-value 7.85x10-8) (S2 Table, full output available online at https://doi.org/10.7488/ds/2631).There was a moderate genetic correlation between the two traits in the GS sample (rG = 0.597, SE = 0.279), and both measures had high genetic correlations with the previously reported findings of Lu et al. (rG = 0.724, SE = 0.312 and rG = 1.021, SE = 0.356 for Horvath-EAA and Hannum-EAA respectively). All the significant SNPs from the Lu et al. analysis of Horvath-EAA had the same direction of effect in GS (S3 Table), with one attaining genome-wide significance (rs143093668, P-value = 3.53x10-8; remaining SNPs had P-values between 5.76x10-2 and 1.34x10-4). Two of the three significant SNPs from Lu et al.'s GWAS of Hannum-EAA had the same direction of effect in GS, although not at genome-wide significance levels in this smaller sample (P-values 1.76x10-3 and 1.75x10-4). Miami plots demonstrating a comparison between the EAA SNP association profiles in the GS and Lu et al. samples are shown in Fig 2 (Horvath-EAA) and Fig 3 (Hannum-EAA). Quantile-quantile plots (QQ plots) for the GWAS of Horvath-EAA and Hannum-EAA in GS are shown in S1 Fig.
.../...
.












