































.
F U L L T E X T S O U R C E : Science Direct
Highlights
• Biological age prediction approaches can be based on data obtained using most omics technologies.
• We shortly discuss main ways of biological age estimation and associated patents.
• We summarize papers on multi-omics approaches in gerontology.
• We describe the top-list of new biomarkers of aging found in the discussed papers for translational purposes.
Abstract
Multi-omics approach nowadays increasingly applied to molecular research in many fields of life sciences. Biogerontology is not an exception; multi-omics gives possibility to evaluate complex biomarkers (or panels) which consist of quantitative as well as phenotypic ones. It is especially important because of weak understanding of the nature of aging. The difficulty now is distinguishing between causes and effects of aging. The application of the whole set of metabolome, methylome, transcriptome, proteome or metagenome data in aging biomarker design becomes the only way to create a holistic view of aging landscape without missing undiscovered mechanisms and levels of organization. We found patents, up-to-date multi-omics datasets and studies, which include bioinformatics innovations to predict biological age in humans. We hope that the review will be also useful for clinicians, because it follows majorly translational purposes.
1. Introduction
We live in an unprecedented period of history. According to the United Nations Prognosis, the number of people in the world aged 60 years or over will be 2.1 billion by 2050 (United Nations, 2017). Together with elevating proportion of elderly people number of chronic aging-related disease cases increases. This will significantly elevate the burden on the healthcare system and economy. Researchers are demanded to develop approaches to improve the performance and quality of life of the elderly.
Aging-related preventive interventions are not possible without personal aging speed measurement. Biomarkers of aging are molecular, cellular or physiological parameters of the body that demonstrate reproducible quantitative or qualitative changes with age. Ideally, interventions should reverse these biomarkers to a younger state or slow down the changes with age (Zhavoronkov et al., 2014). The problem of biomarkers’ identification in the field was postulated in classic works on gerontology (Adelman, 1987; Dean, 1988; Ingram, 1988), but the basis of an approach was built by V. M. Dilman in his elevation hypothesis (1968) and in further neuroendocrine theory of aging, where the hormones played a key role of indicators in homeostasis disorganization during aging (Dil’man and Dean, 1992). Nowadays, hormones (insulin, cortisol, growth hormone, etc.) and other small molecules (glucose, urea, lipids, etc.), associated with aging-related signaling cascades, are the most common biomarkers for a physician, below we discuss the criteria which are essential to sort out an aging biomarker.
The following main criteria for aging biomarker were proposed by Butler et al. (2004):
• Must change with age;
• Have to predict mortality better than chronological age;
• Allow foreseeing the early stages of a specific age-related disease;
• To be minimally invasive - do not require serious intervention or painful procedure.
We extended the list by additional criteria, which could increase the translational potential:
• To be sensitive to early signs of aging (as opposed to frailty and mortality, which are too late for prevention and geroprotection);
• Have predictability with collecting in the foreseeable time range;
• Have low analytical variability (robustness and reproducibility).
While there is no single definitive biomarker for aging, that concise all necessary criteria, a range of different measures have been proposed. The most accessible online database of human aging biomarkers is Digital Aging Atlas (Craig et al., 2015), but this resource have not been updated since 2014, in 2019 the most comprehensive source shedding light on biomarkers in gerontology is a book “Biomarkers of Human Aging” (Moskalev, 2019). In different organs and systems, aging processes occur at different times and at different speeds. Thus, aging biomarker should be multimodal, based on different molecular and physiological parameters.
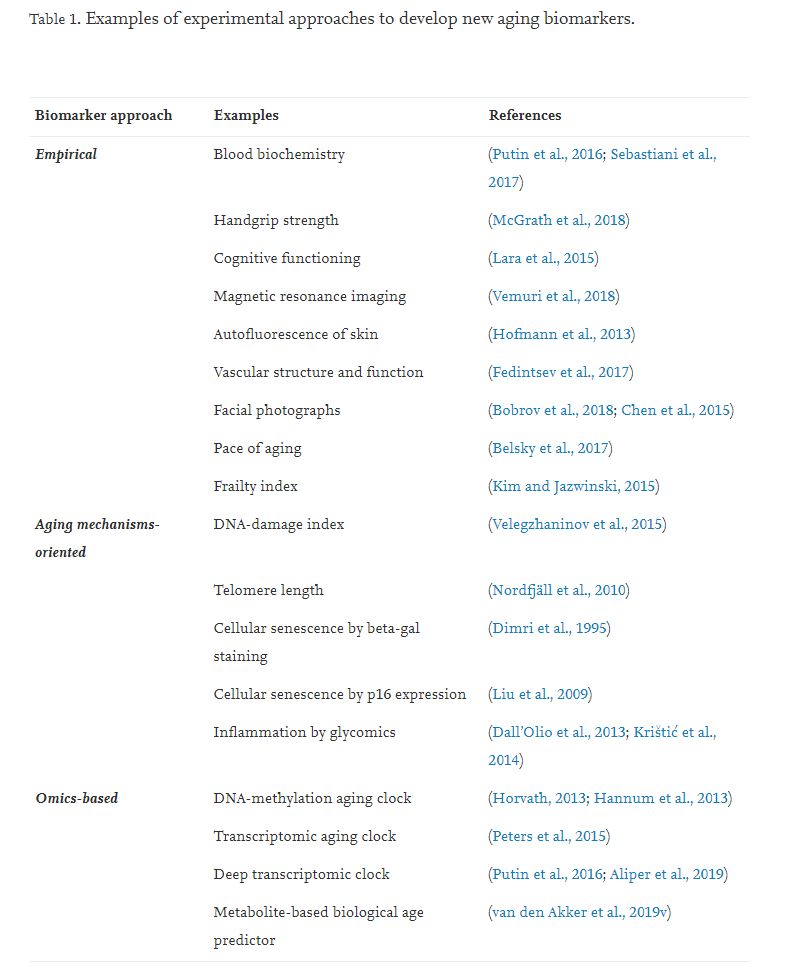
There are three different experimental approaches to develop new aging biomarkers, but it is critical to mention deep transcriptomic clocks separately in the framework of the “Omics-based” measurements (Table 1):
1 Empirical. Search for significant correlations with age among a variety of physiological, psychological, biochemical and other clinical parameters. The advantage of the approach is that the methods have been already used in clinical practice. This approach has maximum translational potential and minimal price, high personal variability and low predictive power.
2 Aging-mechanisms-oriented. Search for predictors of aging among changes associated with known aging mechanisms. Since the approach is based on one of the hypotheses about the causes of aging, it is difficult to confuse the cause with the effect or to base on the false correlation between parameter and age as in the previous one. However, there is always a chance that this is not the main reason of aging. In this case, the variability of the index will be great, and the predictive power will be minimal.
3 Omics. Analysis of age-related correlations among the big data obtained from the analysis of various “omics”: genome, epigenome, transcriptome, metabolome, proteome, microbiome. The main advantage of this approach is that we can assume that we know nothing about the causes of aging at the moment and analyze all possible data of the single person. Deep learning approach. The most up to date and complex way to identify biomarkers of human aging is based on the utilization of deep neural networks which may be trained on any type of appropriate (usually omics) biological data to predict the subject’s age.
Currently, various national projects for biobanking of samples obtained from many people at different ages for subsequent omics analysis exist. The most approximate to aging research omics projects are RNASeq of different tissues of twins (EUROBATS, http://eurobats.eu/),different omics of 3200 subjects (MARK-AGE, (Bürkle et al., 2015)), genomic data of 75,244 participants (UKBiobank, (Pilling et al., 2016)) and multi-omics biobank BBMRI.nl. A multi-omics approach is the most promising due to fast development of the world biobank network, a collection of big data sets, the improvement of bioinformatics methods and artificial intelligence in data analysis (Zhavoronkov et al., 2019). It will allow in the near future to significantly deepen our understanding of aging, and to translate into the clinic the most robust methods for assessing biological age.
Multi-omics approach becomes a golden standard in different fields of bioscience. It gives a deep view of multilayer functional molecular landscape, and it deciphers the complex plexuses of pathways and opens the holistic view of studied processes. Aging as a multifactorial process consequently needs complex approach to be formulated which provides emergence of hidden associations and pathways which may be critical for both geriatric patient and specialist or gerontologist. Multi-omics approach augments the number of obtained markers for biological age measurement and of targets for anti-aging interventions. In the current work we review studies which may impact omics methods common for geriatric practice and fundamental gerontology.
2. Biological vs. chronological age
The usage of chronological age is a common practice in aging studies. But the substrate of aging makes the chronological age an uninformative indicator of aging rate. The existence of premature aging phenomena and tough connection of several chronic diseases with natural manifestations of aging creates a gap between predictive quality of chronological and biological ages. Taking into account the heterochronous origin of aging, the measurement of biological age becomes a complex task based on calculations of numerous target molecules indicating different processes’ dynamics. Sometimes the panels of biomarkers play a role of integrative tools for measurements. Commonly special indexes are used for precise indication of biological age. The MLR (multiple linear regression) approach (used for the cohorts >50 years old, based on the linear correlation of the numerous biomarkers of aging) (Bae et al., 2008; Hollingsworth et al., 1965; Krøll and Saxtrup, 2000); the PCA (Principal component analysis) method unites correlation analysis, redundancy analysis, PCA, and equation construction (Bai et al., 2010; Zhang et al., 2014); Hochschild’s method estimates biomarkers according to their effects on life expectancy (parameters are aggregated into composite validation variables) (Hochschild, 1989); Klemera and Doubal method (KDM) (Klemera and Doubal, 2006) and KDM2 (Cho et al., 2010) uses chronological age as one of the biomarkers, are the most popular ones. The central challenge of biological age estimation: the role of chronological age in different methods of measurement has been still unknown. Some researchers consider it an essential biomarker (Belsky et al., 2015), but others tend to think that chronological age is not required in aging rate measurements (Mitnitski et al., 2017).
The progress in statistics and computational science gives an opportunity to measure biological age in the most rigorous manner using MLR, PCA, Hochschild’s and KDM1/2. Unfortunately, neither of them is completely valid for heterogeneous populations and does not have any clinical determination. Generally, the application of small number of aging biomarkers having different origins results in low resolution of the majority of biomarker panels. The ideal method of biological age estimation must be as comprehensive as possible. Thus, the holistic view is needed. We propose that the creation of the method based on all conceivable multi-omics data as aging biomarkers on integral approach will make the prediction of biological age precise.
The further description of biological age predicting omics approaches will follow the logic of multilayer organization of life.
3. Aging clocks in focus: an overview
3.1. Methylation aging clocks
The term “DNA methylation aging clock” was suggested by Horvath (2013). Nowadays the DNA methylation-based method of biological age estimation has three widely accepted interpretations: Horvath’s, Hannum’s and Levine’s clock (Hannum et al., 2013; Levine et al., 2018). All that methodologies are accurately discussed in Horvath and Raj (2018). Epigenetic clock is a group of “age estimators”, modified CpG islands that are collected all together in one algorithm to estimate biological (epigenetic) age of DNA obtained from any source.
In comparative paper by Liu et al. (2019) it is highlighted that some aging clocks were found to predict age-related pathologies, including diabetes mellitus, coronary heart disease and several cancers. In addition, Liu et al. (2019) add 8 more types of epigenetic clocks: from saliva (Bocklandt et al., 2011), whole blood (Garagnani et al., 2012; Lin et al., 2016; Vidal-Bralo et al., 2016; Weidner et al., 2014; Yang et al., 2016; Zhang et al., 2017) and skin (Horvath et al., 2018). It is worth noting that for every type of epigenetic clock transcriptomic signature was obtained from microarray data (the differential expression of 5028 genes detected in purified monocytes). Interestingly, in Hannum et al. (2013); Horvath (2013); Horvath et al. (2018); Levine et al. (2018); Lin et al. (2016); Yang et al. (2016) epigenetic clocks the transcriptional profiles were very close to each other. The GO Term analysis showed that co-expression modules for all 11 clocks are enriched for metabolic and mitochondrial pathways. The top list of genes associated with 11 epigenetic aging clocks contains IGF1R, SIRT1, NRF2, PIK3R1, ATM, TFAM, NDUFS3, NDUFS7, PHF3, PPP1R12A, USP6, IFIT2, STAT1, NDUFA13, TOMM22, FOXP1, MEF2, ZBRK1, NKX25 and CREB1 (Liu et al., 2019).
It is also important to mention notwithstanding aging clock called DNA methylation GrimAge, this tool is a synthesis of seven DNA methylation surrogates and DNA methylation-associated estimator of smoking status measured in packs per year. This instrument gives a possibility to look at epigenetic aging acceleration from a new side, predicting also the time-to-death (Cox regression P = 2.0E-75) and comorbidity count (P = 7.3E-56), time-to-cancer (P = 1.3E-12), time-to-coronary heart disease (Cox P = 6.2E-24) (Lu et al., 2019). GrimAge is based on the elastic net regression model which sorted out the most critical covariates from candidate biomarkers, the are chronological age (Age), sex (Female), and DNAm based surrogates for smoking pack-years (DNAm PACKYRS), adrenomedullin levels (DNAm ADM), beta-2 microglobulin (DNAm B2M), cystatin C (DNAm Cystatin C), growth differentiation factor 15 (DNAm GDF-15), leptin (DNAm Leptin), plasminogen activation inhibitor 1 (DNAm PAI-1), tissue inhibitor metalloproteinase 1 (DNAm TIMP-1) The linear combination of the covariate values was linearly transformed to be in units of years (Lu et al., 2019).
It is worth noting that in 2019 one more DNA methylation-based biological clock has appeared. The main indicator of biological age in this variant of aging clock is methylation of ribosomal DNA exclusively. The rDNAm age clock models were constructed by application of the elastic net regression algorithm implemented in the glmnet library. This method applies multivariate linear regression with the “predict” and “response” variables being, respectively, the methylation levels of CpGs and the logarithm transformed age. In addition, the model exerts extra constraint on the coefficients of predict variables by adding a penalty to the coefficients using the combination of lasso and ridge regulation methods (Wang and Lemos, 2019). The complex biomarker is evolutionarily conserved. The statement was proved on yeast, Drosophila and human nucleolar DNA. It accurately indicates biological age and reflects organismal response to treatment and potential anti-aging interventions (Wang and Lemos, 2019).
3.2. Transcriptome aging clocks
The transcriptional aging clocks are associated usually with a vast study carried out by Peters et al. (2015) which was based on meta-analysis of 7074 human peripheral blood samples from six independent cohort studies. 11,908 genes were used to create a predictor for age using a leave-one-out-prediction meta-analysis. The average absolute difference between predicted age and chronological age was 7.8 years. A positive increment of age, interpreted as reflecting more rapid biological ageing, was consistently associated with higher systolic and diastolic blood pressure, total cholesterol, high density lipoprotein cholesterol, fasting glucose levels and body mass index. The comparison between this first transcriptomic clock and methylation clocks (Horvath, 2013; Hannum et al., 2013) shows the weaker correlation with chronological age in transcriptional clock, mainly due to the data type, as it was explained by Peters et al. (2015).
One more significant study is dedicated to the transcriptome aging of dermal fibroblasts and a pioneer method of biological age determination for such datasets. The dataset was obtained on 133 people (1–94 years old) and 10 HGPS (Hutchinson-Gilford Progeria Syndrome) patients. Linear discriminant analysis was used for age prediction; it was characterized by a median absolute error (4 years) and a mean absolute error (7.7 years) (Fleischer et al., 2018). This method gives results not far from Horvath’s (Horvath, 2013) or Putin’s (Putin et al., 2016).
Aging speed differs much among individuals and groups; also it may be affected significantly by environmental and hereditary traits. To eliminate the discussed effects a normalized cohort is needed. In paper by Mamoshina et al. (2019) 6465 individuals’ blood samples from 17 datasets were taken. Authors shown that technical performance variations influence blood expression profile more significantly than disease and age itself. After that authors succeeded in batch effect elimination using several normalization techniques (Normalization by Reference Gene, Cross-platform normalization method, Quantile normalization, Distribution Transformation), they also investigated that some of the methods eliminate the age-dependent differences. A deep neural network was utilized as the predictor (0.91 was the accuracy during Pearson correlation and mean absolute error was 6.14 years) (Mamoshina et al., 2019). To conclude, we would like to highlight that transcriptome-based biological age prediction techniques are developing precipitously; their level of accuracy constantly increases, thus, nowadays transcriptomic aging clocks are no worse than methylation clocks which have been already reviewed above.
3.3. Metabolic aging clocks
A paper by Hertel et al. (2016) demonstrates a method of biological age measurement via metabonomics called “the metabolic age score”, applying a nonlinear regression based on urine data obtained by 1H nuclear magnetic resonance spectroscopy. The metabolic age score is predictive for weight loss prognosis made for bariatric surgery patients and is applicable in other fields of personalized medicine.
An innovatively accessible approach of metabolome-based biological age measurement was invented by van der Akker et al. (2019). The vast set of 1H-NMR (Nuclear magnetic resonance) serum metabolomics and phenotypic data including over 25,000 samples derived from 26 community and hospital-based cohorts was used, 56 metabolome endpoints were taken into account. The goal of this study is in accessibility of interactive tools (metaboage.researchlumc.nl) for measurement and high accuracy, which was pronounced by authors, and is expected due to 5-times cross-validation of the model. It evaluates current and predicts cardio-metabolic health, including significant associations with body mass index (P = 2.59E-33), C-reactive protein (p = 1.76E-07), current Type 2 diabetes (P = 5.10 E-17), future cardiovascular events (p = 2.64 E-04) and vascular mortality (p = 8.56E-07). The metabolomics-based age predictor was trained using a linear model (van der Akker et al. 2019).
3.4. Metagenomic data in biological age measurement
Metagenome-based aging clock is a relatively new tool for biological age prediction. The microbiome diversity passes through three basic stages called child’s, adults’ and elderly types, the greatest problem is connected with the absence of adult microbiome concept. The difficulty also appears in seeking human cohorts with similar lifestyles and, additionally, in datasets’ normalization. Generally the biodiversity of human gut microbiota decreases with age, but accidentally, aged individuals demonstrate the same level of biodiversity as adults. The overall microbiome consists mainly of four phyla Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria (Choi et al., 2018). The decrease in abundance of genera Bacteroides, Bifidobacterium, Blautia, Lactobacilli, Ruminococcus is observed during aging, while genera Clostridium, Enterobacteria, Escherichia, Streptococci conversely show growth in number (Claesson et al., 2012; Galkin et al., 2018). Unsurprisingly the aging-associated microbial community studies’ results vary no less than microbial communities. As well as in transcriptomic studies microbiological research seriously depends on methodologies (Woodmansey et al., 2004). There is one statement which is taken as a consensus in translational microbiology of aging: the concentration of short fatty acid producents is lower in gut of aged individuals and it is connected with increased counts of pathogenic and aerotolerant bacteria, which multiplication causes dysbiosis and, as a result, early development of diseases related to age (Galkin et al., 2018). Deep neural network approach was used to count biological age on metagenomic dataset, the list of microbial taxa for search included 1673 names, the mean absolute error is 3.94 years (very close to Horvath’s result which was 3.4 years), with R2 = 0.81 in the variant with the best configuration of a working model, this study was the first case when the biological age was measured on human gut microbial community dataset (Galkin et al., 2018).
3.5. The clinical utility of aging clocks, biomarkers and their role in drug discovery
Commonly the aging clocks and biomarkers are used by academic researchers in the experimental studies, but the popularity of discussed tools in clinicians’ community increases due to the efficacy and cheapness of these instruments, which may accurately draw the dynamic profile of human aging and indicate its pace.
The first known positive result of methylation-clock reversal on human cohort was reported by Fahy et al. (2019). The protocol to regenerate the thymus was utilized during 1 year. The protective immunological changes were described; the risk scores for age-related pathologies were improved seriously and a mean epigenetic age showed a decrease by 1.5 years (-2.5 years compared to the group which was not treated), additionally, the pace of epigenetic aging reversal (taking into consideration the chronological age) was improved from -1.6 year/year (for 0–9 months of treatment) to -6.5 year/year (for 9–12 months of therapy) (Fahy et al., 2019).
It is known that biomarkers are the side outcome objects during drugs’ target search; sometimes the biomarkers and the targets are the same and have very high translational potential (Lopez-Otin et al., 2013). In recent paper by Mitteldorf (2019) methylation age is used as the evaluation of anti-aging interventions and practices, this study with 5000 participants will be used for development of new kind of aging clock which is oriented on synergistic effect detection during age-retarding treatment and also on individual differences. Biological age has already been used as a predictor of mortality in patients with ischemic stroke (Soriano-Tárraga et al., 2018); the approach gives a more trustworthy prognosis than the chronological age does. In addition, biological age of the brain may be utilized in lethality prognosis (Cole et al., 2018). It was reported that methylation age was applied to abuse medicine, thus the increase of biological aging rate was described on patients with alcohol dependence (Rosen et al., 2018). These examples are not numerous but the number of translational research in the area of biological age measurements is developing.
Another segment of geroscience where the biomarkers play a critical role is drug discovery and development. We found a patent on the method of aging biomarker-based skin therapeutic compound discovery (de Oliveira et al., 2019). Biomarker-based design of the study helps to exclude false discovery of therapeutic compounds (Vo et al., 2018). Aging biomarkers may be valuable in Alzheimer's disease drug development and make the final clinical trial phase cheaper. The biomarkers provide the interpretable effects indicating the state of pathophysiological mechanisms standing behind the disease; using non-invasive biomarker-based approach the clinician has an evidence of the disease-modifying effect (Blennow, 2010).
3.6. The intellectual property protection in the area of aging clocks
The attempts to protect the intellectual property in the field of biological age measurement have been made for two decades. One of the earliest documents is “System for improved biological age measurement” (Michaels et al., 2002), the centralized telemedical system which counted the biological age of a patient using several metabolic biomarkers via Internet. The next critical patent on a biological clock was received by Shallenberger (2007): it was based mainly on the measurements of anthropometric, physiological and metabolic scores (Shallenberger, 2007). The most precise epigenetic aging clock was invented in 2014 by S. Horvath and the patent was published in 2016 (Horvath, 2016). It is worth noting that at the same year one more cytosine methylation-based “Method for the determination of biological age in human beings” enriched with hormone and metabolite profiling data was invented and patented (Bürkle et al., 2014). Another epigenetic clock of human skin fibroblasts was patented in 2018, but it is not as precise as Horvath’s one (the error is relatively high: ±5.05 years, compared to ±3.6 in Horvath’s epigenetic aging clock) (Winnefeld et al., 2018). “Deep transcriptomic markers of human biological aging and methods of determining a biological aging clock” is the innovative patented approach in biological age measurement, it contains the information on transcriptome data collection, creation of input vectors, uploading the input vectors in machine learning platform, generation of prediction and automated preparation of the report, it is as precise as Horvath’s aging clock (Aliper et al., 2019).
.../...
F O R T H E R E S T O F T H E S T U D Y, P L E A S E V I S I T T H E S O U R C E .
.
Edited by Engadin, 13 December 2019 - 04:05 PM.















