.
F U L L T E X T S O U R C E : MDPI_Cells
Abstract
In contrast to the programmed nature of development, it is still a matter of debate whether aging is an adaptive and regulated process, or merely a consequence arising from a stochastic accumulation of harmful events that culminate in a global state of reduced fitness, risk for disease acquisition, and death. Similarly unanswered are the questions of whether aging is reversible and can be turned into rejuvenation as well as how aging is distinguishable from and influenced by cellular senescence. With the discovery of beneficial aspects of cellular senescence and evidence of senescence being not limited to replicative cellular states, a redefinition of our comprehension of aging and senescence appears scientifically overdue. Here, we provide a factor-based comparison of current knowledge on aging and senescence, which we converge on four suggested concepts, thereby implementing the newly emerging cellular and molecular aspects of geroconversion and amitosenescence, and the signatures of a genetic state termed genosenium. We also address the possibility of an aging-associated secretory phenotype in analogy to the well-characterized senescence-associated secretory phenotype and delineate the impact of epigenetic regulation in aging and senescence. Future advances will elucidate the biological and molecular fingerprints intrinsic to either process.
1. Introduction
Advancing age has turned into the major risk factor for highly prevalent chronic and devastating diseases, including cancer, cardiovascular and neurodegenerative entities [1,2,3]. Classically, the aging process has been characterized by several candidate hallmarks comprising genomic damage and telomere curtailment, epigenetic alterations, dysregulation of proteostasis and nutrient sensing, mitochondrial dysfunction, stem-cell pool collapse, impaired inter-cell communication and cellular senescence (for a review see [4]). Thus, cellular senescence, first conceived almost 60 years ago and currently defined as a stress response by which cultured cells lose their proliferative capacity in an essentially irreversible manner, is regarded as a direct player in organismal aging in vertebrates [5]. Cellular senescence had been established mainly for replicative human diploid cells grown in vitro [6]. However, since the vast majority of cells in mammals are non-proliferating, the causative impact of replicative senescence in aging is still controversial, and current views are influenced by the relative success of senolytic approaches, which seem to halt and even reverse aging or initiate a state of rejuvenation [7,8]. A second important aspect in this context that emerged only recently as a result of the work of several labs, including our own work, suggests a proliferation-independent senescence-like mechanism in post-mitotic cells including neurons, which might also be involved in the organismal aging process [9,10,11]. Proof of such a phenomenon will conclusively entail the requirement for a revised definition of what cellular senescence subsumes and challenge a hitherto underestimated role of senescence mechanisms in neurodegenerative disorders and other pathobiologies originating from a post-mitotic environment. Thus, consolidation of post-mitotic cell senescence, a concept we term here ‘amitosenescence’ particularly in a neuronal context, would also strongly argue for the as yet controversially discussed functional interdependence of aging and senescence [12]. Proof of amitosenescence as a biological process would further amplify the contribution of cellular senescence to the aging process, in light of the fact that the majority of cells in the organism are nominally post-mitotic, and, in a final consequence, support the claim that aging is at least partially programmed.
In spite of such conceptual dynamics regarding the molecular and cellular landmarks of senescence, and striking new evidence indicating that cellular senescence might play beneficial roles besides tumor suppression, its relevance in healthy and pathological aging as well as in life expectancy is still insufficiently understood. Improvements in this field inevitably will be linked to the establishment of well-accepted, comprehensive and clear definitions and functional stratifications concerning the biological differences between “cell aging” and “cell senescence”. However, both terms are often used indifferently and with low stringency and, thus, can provoke crucial misinterpretations regarding their individual and intercalated roles. Therefore, in the following sections, we will discuss recent and novel concepts on aging and senescence, and report evidence revealing that, although cellular senescence contributes to organismal aging, it appears to be independently regulated in many aspects. In the light of aging being understood as a primarily organismal phenomenon, we will, however, discuss both processes at the cellular level whenever applicable and compare their roles on the basis of factor-specific opposition in terms of conceptual aspects, molecular regulation and biological consequences (for an overview see Figures 1 and 3). By providing such an overview, we aim to achieve a clearer outline of our current scientific definition and understanding of aging and senescence.
2. Current Concepts of Aging and Senescence
The processes driving the progressive decline of functional performance during aging are yet not fully understood. Accordingly, the term ‘aging’ is interpreted ambivalently in the field, and effectors of aging remain a hotly debated issue. Moreover, there is still controversy regarding the interrelation between aging and disease and whether aging reflects a disease state or not [13]. Generally accepted is the basic view that aging subsumes an organismal phenomenon involving an increased chance of dying and/or decreased functionality over time [14]. Although the same definition is applicable at the organ and tissue levels, defining aging at the cellular and molecular levels has been challenging [14].
2.1. Is Aging Programmed?
In the last 50 years, several theories have been proposed to explain the nature and control of aging. Some of them include: 1) the evolutionary theory based on the concepts of deleterious mutational accumulations developed by Peter Medawar in 1952 [15] and antagonistic pleiotropy by George Williams in 1957 [16], which states that specific alleles and mutations that display either positive or no effects on fitness in early development can be detrimental in later life as natural selection fails to remove them, thus causing aging; 2) the free radical theory proposed by Denhan Harman in 1956 [17], suggesting that detriment to cellular components by successive accumulation of oxygen species overwhelms the anti-oxidative mechanisms; 3) the programmed theory proposed by Valter Longo and colleagues in 2005 [18], based on insights by August Weismann in the 19th century, which explains aging and death as a result of a genetic program that evolved to benefit future generations; and 4) the hyperfunction theory stated by Mikhail Blagosklonny in 2008 [19], which suggests that sustained hyperactivity of genes during the reproductive age window causes a state of cellular hypertrophy that results in aging, but is not primarily related to molecular damage.
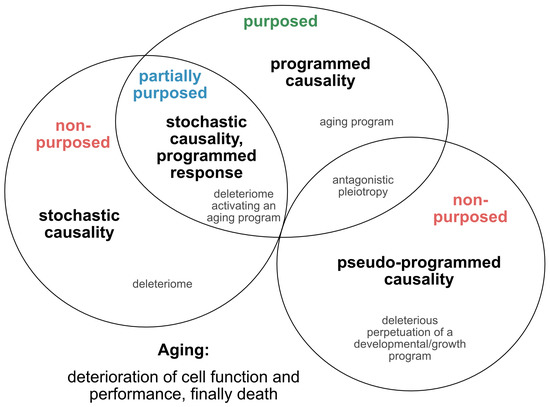
Such approaches, which provide tentative explanations for the causes and mechanisms of the aging process, have continued to evolve and are being translated into novel concepts, fueling the yet ongoing debate over whether aging is programmed, or merely a purposeless, unintended and stochastic accumulation of deleterious events that reduce the performance of an organism (Figure 1). In an attempt to reconcile such theories and pave the way for a directive for promising future concepts, we here extract and re-define the following four essential points of view (Figure 2): 1) the stochastic causative approach propagates the concept that aging arises from the stochastic time-dependent conglomeration of genotoxic events, oxidative stress, loss of repair capacities and other hallmarks that account for cellular deterioration with chronological aging [4]. Whether the development of such a deleteriome is putatively causal to the aging process [20], as already speculated by Harman in 1957, and whether it is devoid of a biological purpose, or follows the evolutionary aim to prioritize young over old organisms to support biological fitness and speed positive selection, are still a matter of debate; 2) the pseudo-programmed causative approach represents the position coined by Mikhail Blagosklonny in 2013, that aging ‘is not and cannot be programmed’ [21], as it reflects the continuation of an embryonic program into adulthood, which cannot be powered off but loses purpose with time. Such a pseudo-program thus shares the same cascades integral to growth and development and drives geroconversion, characterized by growth without cell division, and cellular senescence, the second being regarded as a continuation of differentiation [21]. In conclusion, aging is, therefore, assumed to originate from a genetic pseudo-program that is reminiscent and a relic of a developmental growth program, which, however, is independent from a deleteriome. If such a concept holds true, aging might neither be programmed, nor represent a pure stochastic and random accumulation of events that provoke aging and age-related pathologies, and finally culminate in death. In essence, it mirrors antagonistic pleiotropy, but without the claim of being purposed; 3) the programmed causative approach suggests that aging is partially, or even fundamentally programmed [22]. Likewise, it was recently illustrated that aging might be ruled by a global, unique, but pleiotropic genetic motif.
This highly conserved, transactivating binding motif is strongly associated with transcriptional changes culminating in the process of age-related genetic reprogramming [23,24]. In analogy, reversion of this program, which is essentially nuclear factor kappaB (NF-κB)-mediated, has been illustrated to counteract molecular- and cellular-aging phenotypes [23,24], supporting the notion that the aging process is not self-sustained once initiated by certain genetic mechanisms, but requires a pro-active continuation of a genetic control mechanism. Moreover, the discovery of so-called ‘longevity genes’ and of strategies interfering with, e.g., sirtuins, insulin/insulin-like growth factor-1 (IGF-1) and calorie intake, all of which potently counteract the physiological aging process in different paradigms [25], argues in favor of aging being, at least to some extent, a genetically guided process [23,26,27]. In support of this, longevity factors including Sirtuin 1 (Sirt1) and the Forkhead box protein O (FoxO) are reported to antagonize transcriptional profiles related to the transactivating binding motif mentioned above [26,28,29]. The genetic motif described is also essentially involved in the processes of immuno-aging and age-related inflamm-aging [26,27] and constitutes a main molecular driver of the senescence-associated secretory phenotype (SASP) [30]. Moreover, as it also governs organismal development as a whole [31,32], this concept supports the aforementioned notion of aging representing a ‘quasi-program’ intricately linked to the genetic program underlying growth and development [21]; This motif in terms of NF-κB has recently been highlighted as a downstream target of the cyclic guanosine monophosphate–adenosine monophosphate (GMP–AMP) synthase (cGAS) that controls, in alliance with the class of ‘Stimulator of interferon genes’ (STING), an innate pro-inflammatory immune response elicited by the highly immunogenic nature of ectopic DNA species. In analogy to cancer cells, senescence cells activate the cGAS-STING pathway in response to the occurrence of cytoplasmic heterochromatin, which, in turn, activates paracrine SASP [33,34] and NF-κB that drives itself the SASP and sustains inflamm-aging. Therefore, considering that an innate cGAS-STING immunogenicity path is engaged in the control of senescence might represent a novel direction to counteract senescence. How far hyperactivation of this pathway might bear the risk for tissue damage and chronically sustained inflammation still has to be defined; 4) the stochastic causative, but programmed response approach; based on current knowledge, it is well-accepted that stochastic alterations at the cellular level, e.g., DNA damage, free radical accumulation and mutations, induce compensatory mechanisms to reduce the impact of multiplied damage on an individual’s fitness, health and lifespan. Therefore, lifespan is limited by the capacity to counteract condensed damage and dysfunction. Accordingly, aging will originate from that part of the deleteriome that cannot be neutralized by the compensatory mechanisms individually activated—a process that is initiated with the commencement of life. Counterbalancing mechanisms will work differently in each individual due to variable genetics, epigenetic background, environmental and other stressors and susceptibility factors. One novel aspect among these vulnerability criteria comprises the recent illustration that most organs are composed of an assortment of cells, protein complexes and individual proteins of strikingly different ages, regardless of whether they are quick or slow to regenerate [35,36,37]. Such age mosaicism in the proteome of cells and their structural components assumes exceptional importance in post-mitotic cells as their nucleus will not be replaced over lifetime and thus directly links nucleosome protein turnover to nuclear integrity, chromatin organization and gene transcription. Thus, aging is the failed purpose to counteract damage, and this failure causes functional decline, pathology and death. Factors causative in the initiation of aging are stochastic, but aging-associated response mechanisms such as DNA damage response (DDR), reactive oxygen species (ROS) neutralization, metabolic adaptation, senescence or apoptosis are programmed. This approach also suggests that, although propagation of aging at the organismal level might be irreversible, the kinetics can be decelerated, achieving a partial and ‘relative rejuvenation’ at the cellular, tissue and even organ level.
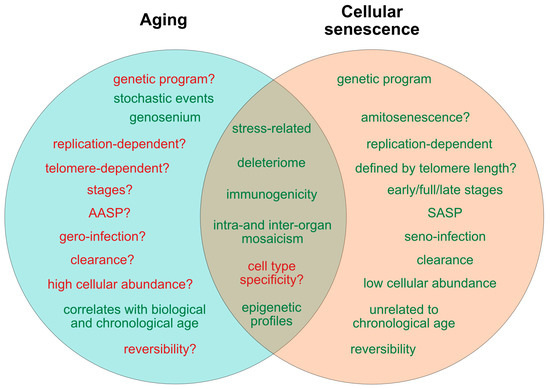
Figure 1. Current determinants and relevant open questions, marking the processes of aging and senescence as discussed in the text. Aspects represented in green are considered as broadly accepted or scientifically consolidated. Novel aspects that are yet unproven, or are under debate, are highlighted in red. SASP = senescence-associated secretory phenotype. AASP = putative aging-associated secretory phenotype as suggested in the text.
Figure 2. Theories on the causality and purpose of aging. Graphically summarized are four contrasting concepts crystallized from current evidence addressing the inductive driving force of aging. Apart from a stochastic deleteriome, there are arguments for a pseudo-programmed, programmed or at least partially programmed nature of aging.
In our view, recent evidence that senescence is based on an unterminated developmental growth program [24] and the finding that the concept of post-mitotic senescence requires the activation of expansion, or ‘growth’ factors as a second hit [38], favor the assumption that aging underlies a grating of genetic determination similarly to what is summarized above under the pseudo-programmed causative approach (Figure 2).
2.2. Single Nucleotide Variants as Genetic Markers Supporting Programmed Aging?
A recent study by Lodato and colleagues, based on a single-cell whole-genome sequencing approach, indicated that in humans terminally differentiated post-mitotic neurons agglomerate somatic mutations with age, thereby displaying age-, region- and disease-specific molecular signatures [39]. Further characterizations of these single nucleotide variants (SNVs) that accumulate almost linearly with aging, a state termed genosenium, and of the even higher abundance of SNVs under neurodegeneration might have the potential to identify genetic markers accessible for therapeutic intervention. Thus, the finding that SNVs and several genes can influence the progression of aging and age-related disorders and impact life expectancy, will surely encourage further unbiased whole genome approaches empowered by enlarged subject numbers. Such studies will putatively relieve the search for a static and universal aging program by the identification of dynamic genetic risk and individualized susceptibility profiles. Such efforts will equally help to better define which concept reflects the nature and biology of aging, and reveal whether there might operate yet undiscovered molecular players, beyond the prolongation of developmental cascades or patterns of SNVs, that drive a pseudo-program of aging. An interesting new step in this direction will be to clarify if such SNVs also support the notion of aging involving genetic fingerprints of growth and development. Accordingly, tracing spontaneous somatic mutations that occur early in life and spread via clonal expansion in replication-competent neural cell entities, as it has been realized by Lee-Six and colleagues in a hematopietic cell context [40] might further improve our understanding of the genetic influence on aging.
2.3. Senescence—A Programmed Process
By contrast, it is accepted that cellular senescence is executed in the framework of sequential and tightly orchestrated stress-induced effector programs involving the down-regulation of Cdks, up-regulation of CdkIs, increased metabolic activity, activated DDR pathways and ultimately the induction of apoptotic effectors [41,42]. Moreover, comparative transcriptome analyses have allowed for the identification of senescence-specific genes including PVRL4, PRODH, LY6D, DAO, EPN3, and GPR172B, all of which are dependent on p53 [43]. In particular, PRODH, DAO, and EPN3 were shown to promote senescence. In contrast, ectopic expression of GPR172B inhibited senescence. The obvious particular role of p53 is supported by a recent computational-assisted work by Kirschner et al. that identified a comprehensive and self-regulatory network of p53-dependent downstream targets, which are specific for acute and chronic kinetics of a cell type-specific p53 regulome [44]. Also, removal of senescent cells under physiological conditions is obviously a programmed process involving both cellular and humoral constituents of the immune system [45].
Despite such a general consensus about senescence underlying a timely and molecularly determined program, the biological importance of the genetic factors engaged is still vague and ill-defined with regard to cellular, tissue and organ specificity. This is in striking contrast to the detailed characterization of other processes such as development and apoptosis that are much better elucidated in terms of kinetics and effector cascades.
2.4. Role of Cell Replication, Telomere Shortening and DNA Integrity for Aging and Senescence
Apart from the controversy on the presence or absence of a putative underlying genetic program connecting aging and senescence, there are several other determinants that draw a picture in favor of aging and senescence being interacting or intercalating, but independent processes (Figure 1 and Figure 3). Senescence was initially described for replicative cells and is still mainly linked to replication-competent cell moieties, whereas there is currently no clear association between the aging process and the turnover rate of cells in different tissues and organs [46,47], a fact that seemingly underlines the independence of the two processes. On the other hand, primarily post-mitotic organs such as the adult brain are equally subjected to age-related dysfunction as replicative organs [48]. Thereby, the presence of a proteome that contains a range of extremely long-lived proteins susceptible to acquiring time-dependent malfunctions might contribute, together with other factors, to explain age-related functional decline in the central nervous system (CNS) and other post-replicative tissues [36,49,50]. Hence, the novel finding of senescence engaging post-mitotic moieties [10], the discovery of patterned SNVs in aged neurons [39], and the identification of inter- and intracellular age differences of individual proteins [35,36,37] suggests that senescence or a senescence-like deregulation is currently underestimated in its impact on aging, and might occur in post-mitotic tissues even at a subcellular scale.
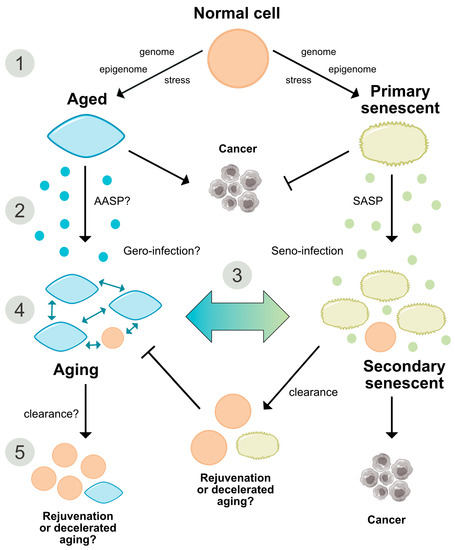
Figure 3. Comparative representation of the aging and senescence processes highlighting different levels of interaction and putative sites of interventions. (1) As discussed in the text, causative mechanisms of aging are still not well understood, however, multiple factors including genetic, epigenetic and stress-related effects seem to have an orchestrated role in the progression of aging. Senescence on the other hand, is seen as a programmed response to different kinds of stressors, which proceed in defined stages. Whether, in analogy, aging also follows a defined program or sequential stages is not known. (2) Senescence involves autocrine and paracrine factors, which are responsible for a ‘seno-infection’ or bystander effect in neighboring cells. There is currently no direct evidence for a similar factor composition propagating the aging process via a kind of ‘gero-infection’. (3) Accumulation of senescent cells has been described as a hallmark of aging; however, whether they are a causative factor or a consequence of tissue and organismal aging is still unknown. As discussed in the text, it appears possible that aging and senescence mutually influence each other through positive feedback at this level, leading to accelerated tissue damage and aging. (4,5) Clearance of senescent or aging cells might constitute putative targets for interventional approaches aimed to reduce or reverse the impact of aging and improve cell and tissue homeostasis by inducing a ‘rejuvenation’ process. SASP = senescence-associated secretory phenotype. AASP = putative aging-associated secretory phenotype as suggested in the text.
Similar considerations apply to the question of how far replication-independent telomeric alterations are relevant for both senescence and aging, and how far replication-associated telomere curtailment propagates either process (Figure 1).
Increased telomere shortening with each cell division cycle is assumed to belong to the key drivers of cellular senescence as soon as a critical length is attained, thereby initiating a DDR entailed by ultimate proliferation arrest [51]. By contrast, naïve telomere length in different species and even inter-individually can greatly vary independently of age, and is generally weakly correlated with their lifespan [52,53]. Therefore, the role of absolute telomere length in aging is still a matter of debate. Alternatively, we recently gathered evidence that the rate of shortening rather than the absolute telomere length might be a better predictor to connect telomeres to the aging process [9]. In analogy, studies performed in proliferation-competent cells emphasize that the main effector responsible for the activation of the p53 and p16Ink4a-Rb pathways involved in telomere-directed replicative senescence is not the loss of telomeric DNA but rather the impaired function of individual telomeres that become extremely short [54,55].
In expansion to the aforesaid in the context of amitosenescence, there is novel evidence that stress-related cellular senescence might be telomere-related also in post-mitotic cells, and that this process plays critical roles during normal aging, and under pathological conditions [10,11,56]. Such a phenomenon appeared even independently from telomere shortening since it could not be reversed by exogenous expression of telomerase [57,58,59]. By contrast, obviously it originates from a DDR activated at deeper sites of damaged telomeres [57,58,59].
.../...
.
Edited by Engadin, 27 January 2020 - 06:15 PM.