































.
Abstract
NF-κB is a transcription factor activated in response to inflammatory, genotoxic and oxidative stress and important for driving senescence and aging. Ataxia-telangiectasia mutated (ATM) kinase, a core component of DNA damage response signaling, activates NF-κB in response to genotoxic and oxidative stress via post-translational modifications. Here we demonstrate that ATM is activated in senescent cells in culture and murine tissues from Ercc1-deficient mouse models of accelerated aging, as well as naturally aged mice. Genetic and pharmacologic inhibition of ATM reduced activation of NF-κB and markers of senescence and the senescence-associated secretory phenotype (SASP) in senescent Ercc1-/- MEFs. Ercc1-/Δ mice heterozygous for Atm have reduced NF-κB activity and cellular senescence, improved function of muscle-derived stem/progenetor cells (MDSPCs) and extended healthspan with reduced age-related pathology especially age-related bone and intervertebral disc pathologies. In addition, treatment of Ercc1-/∆ mice with the ATM inhibitor KU-55933 suppressed markers of senescence and SASP. Taken together, these results demonstrate that the ATM kinase is a major mediator of DNA damage-induced, NF-κB-mediated cellular senescence, stem cell dysfunction and aging and thus represents a therapeutic target to slow the progression of aging.
Introduction
With aging there is an inevitable and progressive loss of the ability of tissues to recover from stress, leading to the increased incidence of chronic degenerative diseases. The loss of tissue homeostasis is driven, in part, by an increase in cellular senescence and a decline in stem cell function, resulting in various aging-related diseases, including osteoporosis, intervertebral disc degeneration, chronic kidney disease, diabetes, neurodegeneration and cancer [1–5]. Cellular senescence, characterized by irreversible cell cycle arrest with sustained metabolic activity, is a biological process that is physiologically required during embryonic development, wound healing and tumor suppression [6–10]. Importantly, senescent cells can develop a senescence-associated secretory phenotype (SASP), including expression of IL-6, IL-1α, IL-1β and TNF-α, that affects neighboring cells, disrupts stem cell niches, alters extracellular matrix, and induces secondary senescence [8, 11, 12]. Cellular senescence is mediated by p53/p21 and p16INK4a/retinoblastoma (Rb) tumor suppressor pathways in response to stress [8, 13] conferred by telomere attrition, DNA damage, oxidative and inflammatory stress and oncogene dysregulation [8].
Cellular senescence directly contributes to the progression of aging. For example, depletion of p16INK4a positive cells in both progeroid and naturally aged transgenic mice expressing an inducible apoptotic transgene from the p16INK4a promoter leads to an extension of healthspan [2, 3]. Similarly, clearance of senescent cells using senolytic agents extends healthspan, improves adult stem cell function and extends lifespan in mice [4, 5, 14–17].
The DNA damage response (DDR), essential for genome stability and organismal survival [18–21], is mediated through pathways that recruit multi-protein complexes to sites of DNA double-strand breaks (DSBs) or stalled replication forks. In particular, the MRE11-RAD50-NBS1 (MRN) complex is recruited to sites of DSBs to facilitate the recruitment, retention and activation of the ataxia-telangiectasia mutated (ATM) kinase [22]. Autophosphorylation of ATM at Ser1981 enhances its kinase activity, leading to phosphorylation of histone H2A variant H2AX (γH2AX) in nucleosomes surrounding damaged sites and recruiting more ATM and other repair factors [22, 23]. Additional DDR proteins, including KRAB-associated protein-1 (KAP1), p53 and checkpoint kinase 2 (CHK2), are phosphorylated by ATM kinase, promoting DNA repair, cell-cycle arrest, apoptosis and/or senescence [24, 25].
The Nuclear Factor κB (NF-κB) family of transcription factors consists of five members in mammalian cells, including RelA (p65), RelB, c-Rel, p50/p105 and p52/p100 [26]. All NF-κB subunits contain a Rel-homology domain (RHD), which is essential for DNA binding activity and dimerization. NF-κB functions to regulate innate and adaptive immune responses, embryonic development, proliferation, apoptosis, oncogenesis and senescence [26]. The canonical NF-κB pathway is activated by inflammatory stimuli, such as TNF-α, IL-1β and LPS, which lead to the activation of the IκB kinase (IKK). IKK is composed of two catalytic subunits, IKKα and IKKβ, and one regulatory subunit, NF-κB essential modifier (NEMO) or IKKγ [26]. Activated IKK complex phosphorylates the inhibitory protein IκBα to facilitate its polyubiquitination and degradation by the 26S proteosome, resulting in the translocation of the NF-κB heterodimer into the nucleus where it regulates gene transcription [26]. In addition, genotoxic stress activates a TNF-α-independent, but ATM-dependent NF-κB pathway via nuclear-localized NEMO [27, 28]. ATM phosphorylates NEMO at Ser85, which in turn induces sumoylation and mono-ubiquitination of NEMO at Lys277 and 309 [29]. These post-translational modifications eventually lead to the nuclear export of the ATM-NEMO complex to the cytoplasm where it associates with ubiquitin and SUMO-1 modified RIP1 and TAK1, activating the catalytic IKKβ subunit [28, 30].
NF-κB activity increases in multiple tissues of humans and rodents with aging and promotes cellular senescence [31–36]. Genetic depletion of RelA/p65 in aged mouse skin and a mouse model of human progeroid syndrome, reversed gene expression signature of aging and aging phenotypes [35, 37]. In addition, heterozygosity of p65/RelA in a mouse model of Hutchinson-Gilford Progeria Syndrome (Zmpste24-/-) resulted in attenuated aging pathology and a prolonged lifespan, linked in part to a reduced systemic inflammatory response and a reduction in ATM/NEMO-mediated NF-κB activation [34]. In addition, Nfkb1-/- (p50-/-) mice have increased low-grade inflammation with signs of premature aging, including neural degeneration, impaired regeneration and declined overall lifespan [38–41]. Activation of NF-κB also is associated with multiple aging-related chronic diseases, including Alzheimer’s disease, Parkinson’s disease, Type II diabetes, osteoporosis and atherosclerosis [42], possibly through an increase in secretion of SASP factors [43].
DNA damage is known to increase with aging as demonstrated by an increase in DNA damage foci (γH2AX) and oxidative DNA lesions (8,5’-cyclopurines) [44, 45]. Intriguingly, persistent DDR signaling mediated by ATM activation has been reported to contribute to cellular senescence and SASP [46]. In vitro, SASP is dependent on ATM activation, suggesting a molecular link between ATM and NF-κB [8, 46, 47]. However, it is still unclear if aberrant DNA damage-induced activation of ATM in vivo exacerbates the cellular stress response to increase NF-κB, senescence, SASP and subsequently aging.
To address the role of ATM in driving NF-κB mediated senescence and aging, we used Ercc1-/Δ mice that model a human progeroid syndrome caused by impaired repair of DNA damage. The mice express only 5% of the normal level of the DNA repair endonuclease ERCC1-XPF that is required for nucleotide excision, interstrand crosslink and repair of some double-strand breaks. As a consequence, the Ercc1-/Δ mice spontaneously and rapidly develop progressive age-related diseases, including osteoporosis, sarcopenia, intervertebral disc degeneration, glomerulonephropathy, neurodegeneration, peripheral neuropathy and loss of cognition [48].
Here, we demonstrate that ATM and downstream effectors are persistently elevated in Ercc1-/∆ and naturally aged mice, concomitant with hyperactive NF-κB signaling. Reducing ATM activity either genetically or pharmacologically reduced cellular senescence and downregulated NF-κB activation in cell culture. Importantly, Ercc1-/Δ mice heterozygous for Atm exhibited significantly reduced NF-κΒ activity, reduced cellular senescence, improved muscle-derived stem/progenitor cell function and attenuated age-related bone and intervertebral disc pathologies, leading to an extension of healthspan. Similarly, inhibiting ATM in Ercc1-/∆ mice by treatment with the ATM inhibitor KU-55933 reduced senescence and SASP marker expression. These results demonstrate a key role for ATM in aging and suggest that it is a therapeutic target for delaying or improving numerous age-related diseases.
Results
NF-κB and ATM signaling are highly activated in cellular senescence, as well as accelerated and natural aging
Our previous studies using transgenic mice carrying a NF-κB-dependent EGFP reporter demonstrated an increase in the percentage of EGFP-positive cells in the liver, kidney, skeletal muscle and pancreas of progeroid Ercc1-/Δ and aged wild-type (WT) mice [35]. To further quantify NF-κB activation with aging, p-p65 (Ser536), a marker of NF-κB activation, was measured in murine liver (Figure 1A). Phosphorylation of p65 was significantly increased in 16-week-old Ercc1-/Δ mice compared to age-matched WT mice (Supplementary Figure 1A). In addition, there was an increase in the level of p-ATM as well as two senescence markers, γH2AX [49] and p21, in Ercc1-/∆ liver compared to WT controls (Figure 1B and Supplementary Figure 1B). To determine if NF-κB and ATM were activated in WT mice with aging, p-p65, p-IκBα and p-ATM were measured by immunoblot in liver extracts from WT mice at multiple ages. The levels of p-p65 and p-IκBα increased gradually with age from 3 to 12 and 24-months of age (Figure 1C and Supplementary Figure 1C). These correlated with increased levels of p-ATM and the senescence marker p21 at 12 and 24 months of age (Figure 1D and Supplementary Figure 1D).
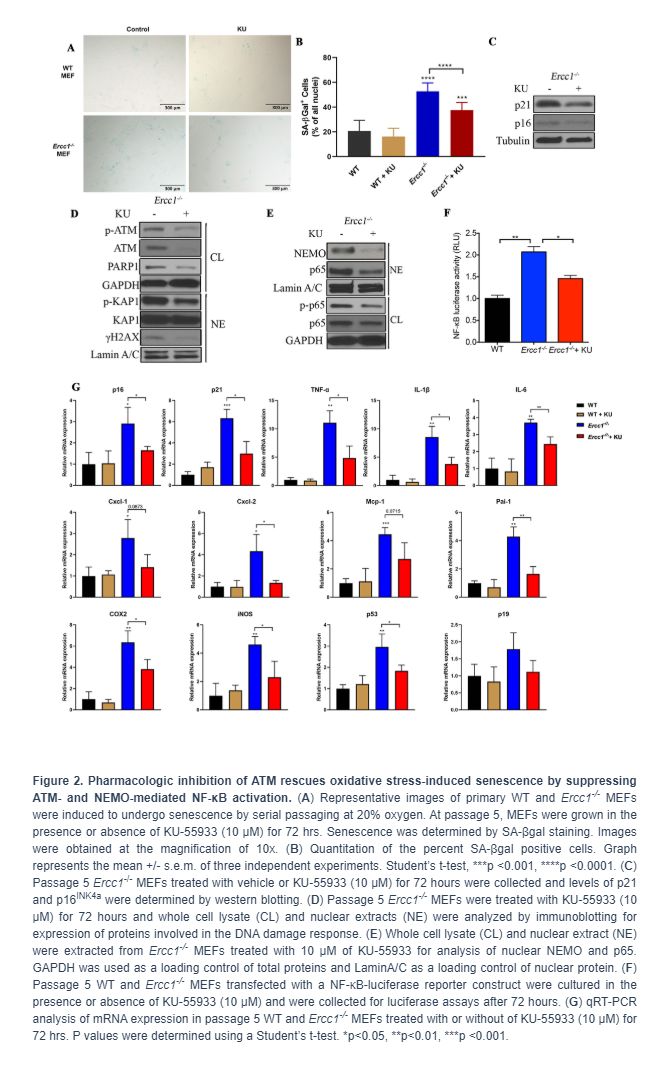
Genetic depletion of Atm decreases genotoxic stress-induced cellular senescence
To eliminate possible off-target effects of the ATM inhibitor KU-55933, Ercc1-/- MEFs heterozygous for Atm (Ercc1-/-Atm+/-) were generated. The Ercc1-/-Atm+/- MEFs had increased proliferation compared to Ercc1-/- MEFs (Figure 3A). There also was a reduction in the percent of SA-ßgal+Ercc1-/-Atm+/- MEFs compared to Ercc1-/- MEFs (Figure 3B and 3C). Expression of p16INK4a also was reduced in the double mutant MEFs (Figure 3D). Finally, there was a reduction in the level of secreted IL-6, a SASP factor, in conditioned media from Ercc1-/-Atm+/- MEFs compared to Ercc1-/- cells (Figure 3E). Taken together, these results suggest that ATM promotes genotoxic stress-induced cellular senescence, in part, through the activation NF-κB signaling.
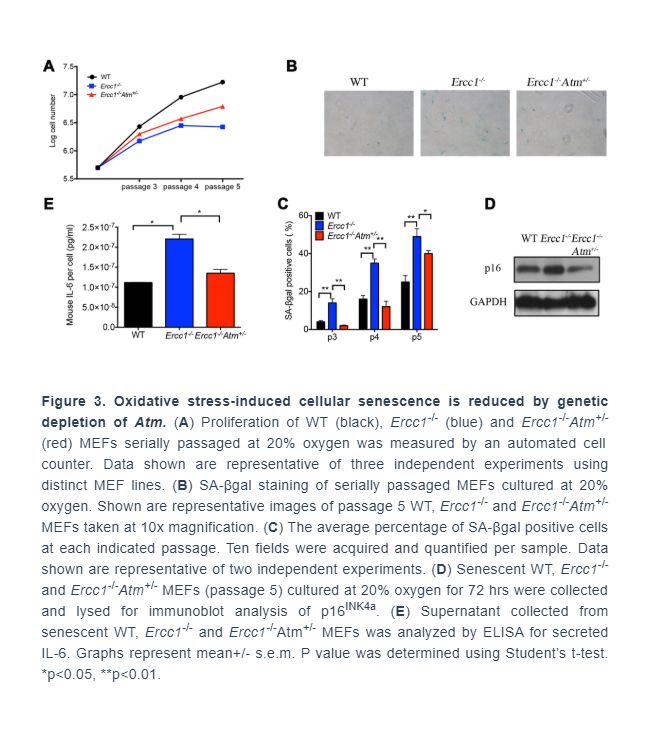
Genetic depletion of Atm extends healthspan in Ercc1-/Δ mice by reducing cellular senescence
To determine if Atm heterozygosity extends healthspan in Ercc1-/∆ mice, age-related symptoms, including kyphosis, tremor, ataxia, gait disorder, hind limb muscle wasting, forelimb grip strength and, in particular, dystonia were measured weekly in Ercc1-/∆ and Ercc1-/∆Atm+/- mice. As shown in Figure 4A and 4B, Atm heterozygosity reduced the severity and slowed progression of aging symptoms in Ercc1-/Δ mice.
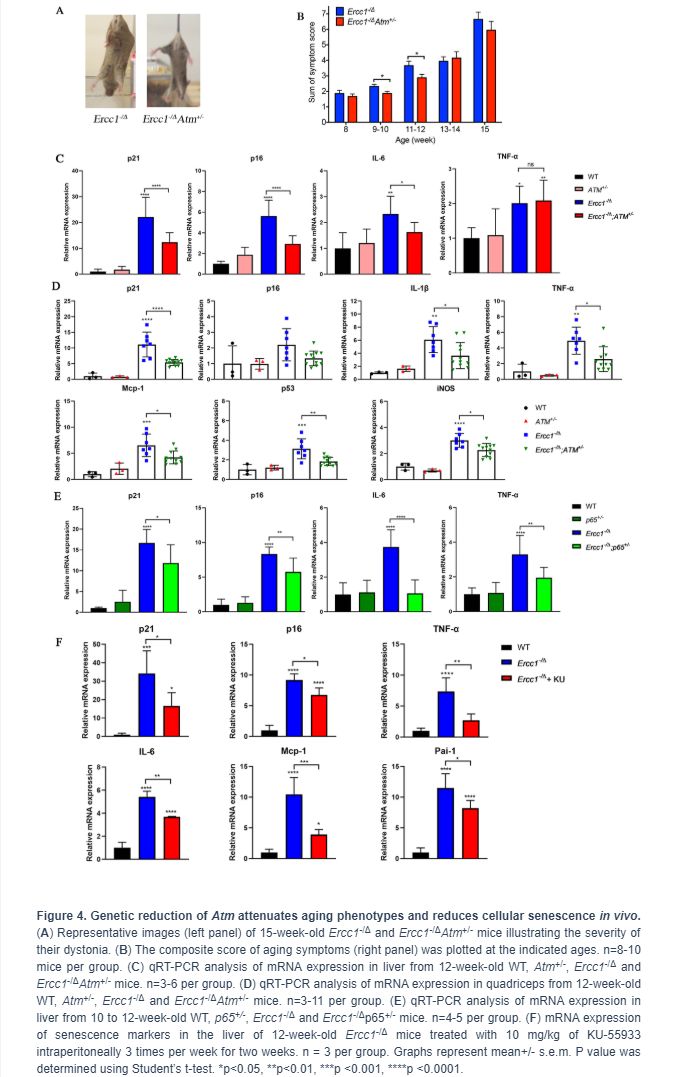
To determine if the extended healthspan correlated with reduced cellular senescence in Ercc1-/∆Atm+/- mice, the levels of expression of senescent markers and SASP factors were measured by qRT-PCR analysis. Expression of p21 was significantly reduced in 12-week-old Ercc1-/∆Atm+/- livers compared to those from aged-matched Ercc1-/∆mice, as was IL-6, a SASP factor and NF-κB target gene (Figure 4C). Similarly, the expression of senescence markers and SASP factors (except p16) were significantly reduced in Ercc1- /∆Atm+/- quadriceps compared with those in Ercc1-/∆mice (Figure 4D). In wildtype mice, there was no effect of ATM heterozygosity on expression of these senescence and SASP markers. Interestingly, IL-6 and TNF-α, but not p21Cip1 or p16INK4a expression, were significantly lower in liver of 12-week-old Ercc1-/Δ p65+/- mice compared to age-matched Ercc1-/ΔAtm+/- mice (Figure 4E). This indicates a distinct role for NF-κB in promoting aging. Taken together, these results suggest that genetic reduction of ATM leads to a reduction in cellular senescence and aging symptoms in vivo.
To determine if pharmacologic inhibition of ATM in Ercc1-/∆ mice confers a similar reduction in senescence and SASP markers as ATM heterozygosity, Ercc1-/∆ mice were treated three times per week i.p. for 2 weeks with 10 mg/kg of KU-55933. The mice were then analyzed for the extent of senescence and SASP in different tissues. As shown in Figure 4F, the levels of expression of senescent markers and SASP factors were measured were significantly reduced in liver tissues from Ercc1-/∆mice compared to untreated controls.
Atm haploinsufficiency improves function of muscle-derived stem/progenitor cells (MDSPC)
A decline in stem cell function has long been associated with aging, which in the musculoskeletal system leads to sarcopenia and muscle wasting [1, 52]. We previously reported that muscle-derived stem/progenitor cells (MDSPCs) isolated from Ercc1-/Δ mice failed to proliferate or differentiate properly, similar to MDSCPs from naturally aged mice [53]. Interestingly, the function of MDSPCs isolated from Ercc1-/∆ mice was partially restored by Atm heterozygosity (Figure 5). In fact, MDSPCs isolated from Ercc1-/∆Atm+/- mice showed similar levels of myogenic differentiation to WT mice (Figure 5A, 5C and 5D). Proliferation of MDSPCs isolated from Ercc1-/∆Atm+/- mice also increased by 50% compared to control MDSPCs (Figure 5B). This result is consistent with delayed onset and reduced severity of gait disorder observed in Ercc1-/ΔAtm+/- mice, which is partly due to severe muscle wasting (data not shown).
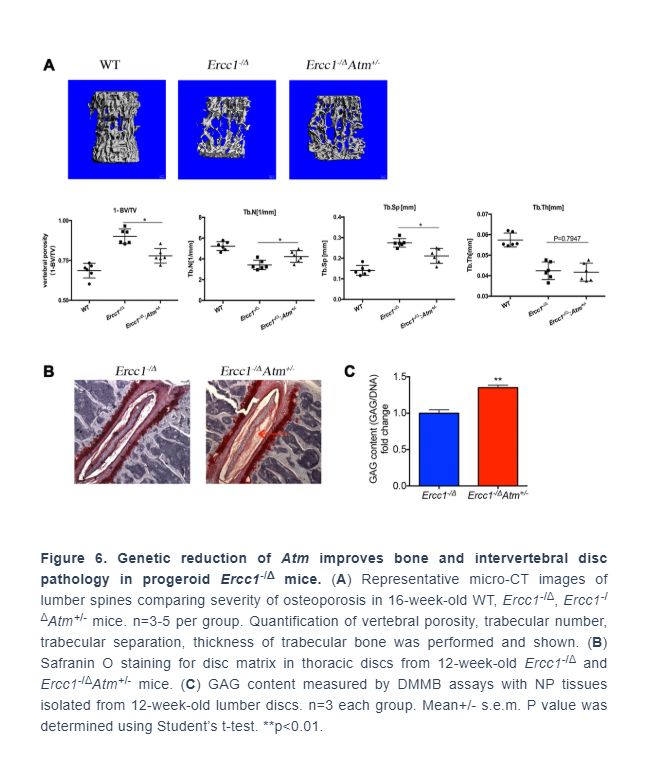
.../...
.














