































.
Abstract
Replication Stress (RS) is a type of DNA damage generated at the replication fork, characterized by single-stranded DNA (ssDNA) accumulation, and which can be caused by a variety of factors. Previous studies have reported elevated RS levels in aged cells. In addition, mouse models with a deficient RS response show accelerated aging. However, the relevance of endogenous or physiological RS, compared to other sources of genomic instability, for the normal onset of aging is unknown. We have performed long term survival studies of transgenic mice with extra copies of the Chk1 and/or Rrm2 genes, which we previously showed extend the lifespan of a progeroid ATR-hypomorphic model suffering from high levels of RS. In contrast to their effect in the context of progeria, the lifespan of Chk1, Rrm2 and Chk1/Rrm2 transgenic mice was similar to WT littermates in physiological settings. Most mice studied died due to tumors -mainly lymphomas- irrespective of their genetic background. Interestingly, a higher but not statistically significant percentage of transgenic mice developed tumors compared to WT mice. Our results indicate that supraphysiological protection from RS does not extend lifespan, indicating that RS may not be a relevant source of genomic instability on the onset of normal aging.
Introduction
For decades, researchers have investigated the process of aging: a decline in health over time, which is generally considered to be due to an accumulation of cellular damage [1, 2]. This cellular damage has different causes, but genomic instability is considered one of the main factors that contribute to cellular aging [3]. Genetic damage such as point mutations, chromosomal rearrangements, aneuploidy and copy number variations accumulate during life and are caused by endogenous and exogenous sources, including UV radiation, ionizing radiation and reactive oxygen species (ROS) [3, 4]. While the accumulation of genomic instability over time can cause cellular senescence and apoptosis in the case of aging, in other cases it can lead to uncontrolled cellular proliferation, and is therefore associated with an increased cancer risk [3, 4].
Defects in DNA repair proteins lead to an accumulation of DNA damage and thus contribute to accelerated aging, as is the case for several human diseases including Cockayne, Bloom and Werner syndromes, trichothiodystrophy and ataxia telangiectasia [5, 6]. In addition, several mouse models have confirmed that mutations in DNA repair proteins lead to accelerated aging. These mice exhibit accelerated aging and aging-related phenotypes such as alopecia, grey hair, osteoporosis, cachexia, neurological abnormalities, retinal degeneration and a predisposition to a wide variety of cancers [5, 7–12]. Taken together, this indicates that an exacerbated accumulation of DNA damage leads to premature aging, albeit the actual contribution of DNA damage to normal aging remains to be elucidated. More importantly, which types of DNA damage play a central role in the context of aging is still unknown.
Besides strategies to accelerate aging, genetic manipulation studies in worms, flies and mice have also successfully lead to increases in lifespan, suggesting a direct implication of the manipulated genes in normal aging [1, 13]. For instance, mice overexpressing the spindle assembly checkpoint protein BubR1 show a reduction in age-dependent aneuploidy, reduced incidence of cancer, and an increased healthy lifespan compared to WT mice [14]. Another relevant mouse model with delayed aging is a mouse model with extra copies of tumor suppressor genes Trp53 and its positive regulator Arf, which in addition to having an increased median lifespan has a reduced tumor incidence [15]. The median survival is increased further in mice with constitutive telomerase reverse transcriptase (TERT) overexpression in addition to extra copies of Trp53 and Arf [16].
In recent years, replication stress (RS) has been acknowledged as an important source of endogenous DNA damage [17]. RS is a type of DNA damage that occurs when obstacles to replication lead to an accumulation of single stranded DNA (ssDNA) at stalled replication forks, which is recognized by ssDNA binding protein RPA. This initiates a signaling cascade involving Ataxia Telangiectasia and Rad3-related (ATR) kinase and CHK1 which promotes DNA repair, cell cycle arrest, and apoptosis [18–20]. Similar to other types of DNA damage, RS has been linked to aging. For instance, aged hematopoietic stem cells (HSCs) exhibit increased levels of RS compared to young HSCs [21]. In addition, mutations in the ATR gene cause Seckel syndrome in humans, which is characterized by progeria, growth retardation, microcephaly, mental retardation and dwarfism [22] (OMIM210600). The involvement of RS in premature aging has also been shown experimentally with a mouse model for Seckel syndrome [12]. ATR-Seckel mice exhibit a phenotype similar to that of human patients, which is further aggravated in combination with several cancer-driving mutations such as the Myc oncogene or the absence of the tumor suppressor p53 [12, 23]. ATR-Seckel mice show high levels of RS during embryonic development, accelerated aging in adult life and early lethality [12]. Interestingly, mice harbouring extra alleles of Chk1 (Chk1Tg) or of the ribonucleotide reductase (RNR) regulatory subunit Rrm2 (Rrm2Tg), which is a limiting factor for dNTP production, improved the lifespan and alleviated the progeroid phenotype of ATR mutant mice [24, 25]. These Chk1 and Rrm2 transgenic mice carry bacterial artificial chromosome (BAC) alleles of the respective genes, including exons and introns, under their own endogenous promoters. This strategy provides supraphysiological levels of CHK1 and RRM2 while preventing overexpression in tissues where these genes are normally not expressed, and was proven successful with the Trp53 BAC-transgenic mouse model [26]. Collectively, these studies suggested that RS might have important implications in mammalian aging. However, the effect of Chk1 and Rrm2 expression levels on normal aging, in mice with physiological levels of ATR, remains to be elucidated.
In the current study, we investigated the effect of supraphysiological levels of CHK1 and RRM2, which confer extra protection against RS, on normal aging. We utilized cohorts of WT, Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg mice to asses tumor-free survival of these mice. We found no differences in survival between the genotypes and all mice exhibited similar signs of aging, although there was a higher tumor incidence in the transgenic mice compared to WT mice. Thus, supraphysiological levels of CHK1 and RRM2 do not affect normal aging in mice.
Results
Generation of mice with supraphysiological levels of CHK1 and RRM2
In order to investigate the effect of RS on lifespan, we used the Chk1Tg and Rrm2Tg mouse models previously generated in our laboratory [24, 25]. Chk1Tg and Rrm2Tg mice were crossed in order to obtain Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg mice. Transgenic mice were not phenotypically distinguishable from WT littermates and were born in accordance with Mendelian ratios (Chi-square p-value = 0.243) (Table 1).
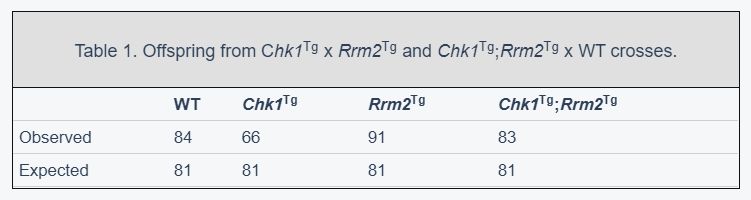
MEFs with increased levels of RRM2 and CHK1 show less damage after induction of RS
To confirm that supraphysiological levels of CHK1 and RRM2 could protect cells against RS, we generated mouse embryonic fibroblasts (MEFs) from crosses between Chk1Tg and Rrm2Tg mice. Chk1Tg and Rrm2Tg MEFs have been previously described to be resistant to RS induced by the ribonucleotide reductase inhibitor hydroxyurea (HU) [24, 25]. Genomic PCR genotyping confirmed the four different MEF genotypes: WT, Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg (Figure 1A). In addition, increased protein levels of RRM2 and CHK1 in MEFs carrying the Rrm2 or Chk1 transgene were confirmed by Western blotting (Figure 1B).
We then assessed whether elevated levels of CHK1 and RRM2 would influence cell proliferation, and found that Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg MEFs proliferated similarly to WT MEFs (Figure 1C). Similarly, Edu incorporation analyses showed similar cell cycle distribution and percentage of replicating cells in all the genoytypes (Figure 1D and Supplementary Figure 1).
Next, we assessed whether MEFs carrying extra copies of Rrm2 and Chk1 were protected against RS by assessing γH2AX levels in these cells. Using high-content microscopy, we found that Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg MEFs have lower basal levels of γH2AX compared to WT MEFs (Figure 1E, 1F and Supplementary Figure 2A, 2B). In addition to assessing γH2AX levels in unstressed conditions, we induced RS by treating the cells with CHK1-inhibitor UCN-01 at different doses and found that the cells with higher CHK1 and/or RRM2 levels showed a decrease in γH2AX intensity compared to WT MEFs (Figure 1E and Supplementary Figure 2A). In addition, the MEFs were treated with HU (Figure 1F and Supplementary Figure 2B). In accordance with previously published data on Chk1 and Rrm2 transgenic MEFs [24, 25], we observed lower γH2AX intensity in these MEFs compared to WT. Importantly, these γH2AX analyses were performed in early passage MEFs (passage 3), when replication and cell proliferation were proficient and comparable among the different genotypes. Thus, the differences found in γH2AX cannot be explained by differences in replication or proliferation rates. These data confirm that cells from mice carrying extra copies of the Rrm2 or Chk1 genes show less DNA damage after induction of RS with HU and UCN-01 compared to cells from WT mice.
Supraphysiological levels of CHK1 and RRM2 do not influence lifespan in mice
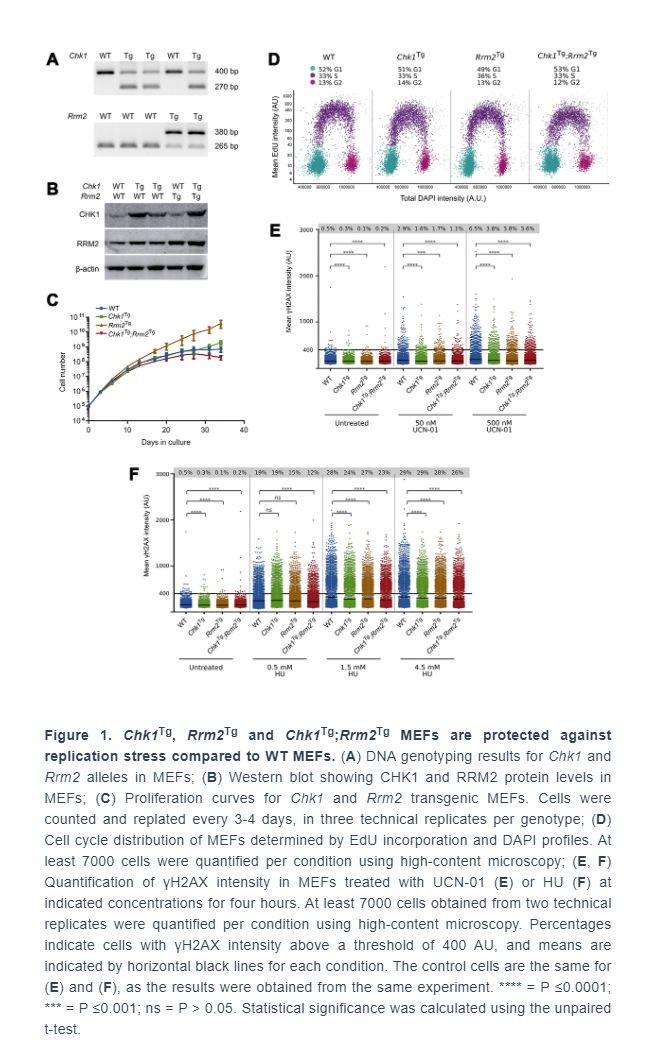
Next, we aimed to investigate whether the protection against RS conferred by extra copies of Chk1 and Rrm2 would be reflected in the survival of mice, as they did in the context of reduced levels of ATR [24, 25]. To this end, we used mice containing the Chk1 and/or Rrm2 transgenes, and assessed tumor-free survival of these mice. Mice were euthanized when they had noticeable tumors or were visibly ill, as observed by rapid weight loss, hunched posture, rough hair coat, labored breathing, lethargy, impaired mobility or abdominal swelling. Only female mice were included in the cohorts, since they could be group-housed more easily than males.
Our results show that survival was not increased due to CHK1 or RRM2 levels: the survival of WT, Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg was not significantly different (log-rank p=0.3944) (Figure 2A). We collected tissues of two year-old mice (not included in survival curve). H&E staining of spleens, livers and kidneys of mice did not show obvious differences between the genotypes (Figure 2B). Furthermore, we observed no noticeable differences in appearance among the mice with different genotypes, as reflected by their weight at 1 year of age (Figure 2C). In addition, all mice displayed comparable signs of aging such as weight loss, grey hair and baldness (data not shown). In summary, unlike in an ATR-deficient background, extra copies of Chk1 and Rrm2 did not increase the overall survival of mice.
Discussion
In the presented study, we aimed to determine whether overexpression of Chk1 and/or Rrm2 could extend mammalian lifespan by utilizing transgenic mouse models carrying extra copies of the Chk1 and Rrm2 genes. These BAC-transgenic mouse models contain extra copies of the genes of interest, which keep the expression of the genes in a physiological range and maintain their endogenous regulation and has been a successful strategy in the past [15, 26]. Previous studies have shown that defects in RS-related proteins lead to increased RS levels and accelerated aging [12, 30, 31]. Lopez-Otin and colleagues described in 2013 the hallmarks of aging [3], one of them being DNA damage. However, a hallmark of aging should not only cause accelerated aging when aggravated, but also increase lifespan when attenuated [3].
Previous studies from our group showed that CHK1- and RRM2-overexpressing MEFs have lower levels of DNA damage in basal conditions and have greater protection against HU- and UCN-01-induced RS [24, 25], and we hypothesized that this protection against RS could lead to an increase in lifespan in vivo. However, we did not observe a difference in lifespan between Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg and WT mice, and all mice developed similar age-related symptoms. Based on these results we conclude that supraphysiological levels of CHK1 and RRM2 do not influence normal aging, and more generally, we propose that physiological RS might not be an important driver of aging. However, we cannot rule out that RS might influence aging in a less controlled environment where mice could be exposed to pathogens and other insults. Although Chk1Tg and Rrm2Tg mice were previously shown to extend the lifespan and alleviate the progeroid symptoms of ATR-deficient mice [24, 25], this is not a direct indication that CHK1 and RRM2 levels influence normal aging. CHK1 overexpression could compensate for ATR deficiency, as CHK1 is a downstream target of ATR. For RRM2 this connection to the ATR pathway is not as direct, although studies in yeast have shown that ATR ortholog Mec1 can activate the ribonucleotide reductase (RNR) complex [32, 33]. Therefore, extra levels of RRM2 could also partially compensate for ATR deficiency.
Interestingly, a higher fraction of the Chk1 and Rrm2 transgenic mice developed tumors, primarily lymphomas, compared to WT mice, although this difference was not statistically significant. The exact role of the RS response in regard to tumorigenesis is complex [34, 35]. On the one hand, CHK1 is known as a tumor suppressor, and its expression can induce senescence and apoptosis, thereby limiting tumorigenesis [36, 37]. On the other hand, experiments with Chk1Tg MEFs have shown that overexpression of CHK1 can promote oncogenic transformation, and elevated CHK1 levels are present in lymphomas, suggesting that CHK1 could also have a role in promoting tumorigenesis [25, 38]. In relation to RRM2, nucleoside supplementation has been shown to both limit and promote transformation [39, 40]. In addition, increased nucleotide levels can lead to errors during DNA replication and tumorigenesis [27, 41]. Our findings favor the concept that RS is not a requirement for transformation, but also that protection against RS does not prevent tumorigenesis.
Furthermore, the increased tumor incidence in Chk1 and Rrm2 transgenic mice could affect their survival, as is the case for TERT-overexpressing mice. Several Tert transgenic mouse models have an increased incidence of spontaneous tumors [42–45]. Thus, extension of lifespan by TERT-overexpression is only possible in the context of a cancer-resistant background [16]. Since we observed a slightly higher tumor incidence in Chk1 and Rrm2 transgenic mice, these mice could possibly survive longer in a cancer-resistant background. Also, the process of aging is complex and influenced by different factors. Conversely, overexpression of DNA damage proteins may improve several aspects of aging, but not the aging process as a whole, and therefore we cannot rule out the involvement of RS on aging. Nevertheless, our data indicate that RS might not be a relevant contributing factor to normal aging in mice.
.../...
.















