.
O P E N A C C E S S S O U R C E : nature
Abstract
Because old age is associated with defects in circadian rhythm, loss of circadian regulation is thought to be pathogenic and contribute to mortality. We show instead that loss of specific circadian clock components Period (Per) and Timeless (Tim) in male Drosophila significantly extends lifespan. This lifespan extension is not mediated by canonical diet-restriction longevity pathways but is due to altered cellular respiration via increased mitochondrial uncoupling. Lifespan extension of per mutants depends on mitochondrial uncoupling in the intestine. Moreover, upregulated uncoupling protein UCP4C in intestinal stem cells and enteroblasts is sufficient to extend lifespan and preserve proliferative homeostasis in the gut with age. Consistent with inducing a metabolic state that prevents overproliferation, mitochondrial uncoupling drugs also extend lifespan and inhibit intestinal stem cell overproliferation due to aging or even tumorigenesis. These results demonstrate that circadian-regulated intestinal mitochondrial uncoupling controls longevity in Drosophila and suggest a new potential anti-aging therapeutic target.
Introduction
Most animals exhibit behavior and physiologies linked to a 24-h circadian rhythm, controlled by endogenous circadian clocks. These clocks are composed of transcriptional feedback loops regulating the expression of genes controlling diverse cellular functions. In Drosophila, the circadian transcriptional activators Clock and Cycle induce the oscillating expression of hundreds of target genes, including period (per) and timeless (tim), which encode repressors of Clock and Cycle activity (Fig. 1a). Because organisms lose circadian rhythmicity with age, it has been hypothesized that loss of circadian regulation contributes to aging and limits lifespan. Specifically, there has been interest in the impact of circadian-regulated metabolism on lifespan and aging, as many mechanisms regulating organismal lifespan involve large metabolic changes, including circadian-regulated metabolic genes1,2. Most reports investigating core molecular clock components and lifespan examined the circadian transcriptional activators (i.e., Clock or Cycle) and found that disruption of these activators led to metabolic dysfunction and shortened lifespan3,4,5,6,7. Studies examining loss of circadian transcriptional repressors (i.e. Period, Timeless, or Cryptochrome) and the impact on metabolism, healthspan, and lifespan have been more controversial5,8,9,10,11,12. While a number of studies identified the detrimental effects of loss of the transcriptional repressor per2 in the mouse13,14 or per in Drosophila9,15,16, loss of per2 may also have beneficial effects on specific aspects of healthspan and lifespan12,17,18,19. For example, loss of per2 in the whole mouse has been associated with increased metabolic rate, lowered fat storage, increased leptin levels, and decreased insulin resistance compared with control animals, indicating a possibly favorable metabolic state under ad libitum feeding conditions17,20. Thus, while loss of circadian regulation has pathological effects, loss of circadian regulation in specific genetic and environmental contexts may have metabolic advantages. The specific mechanisms underlying circadian-regulated metabolism and their roles in aging and longevity remain unclear.
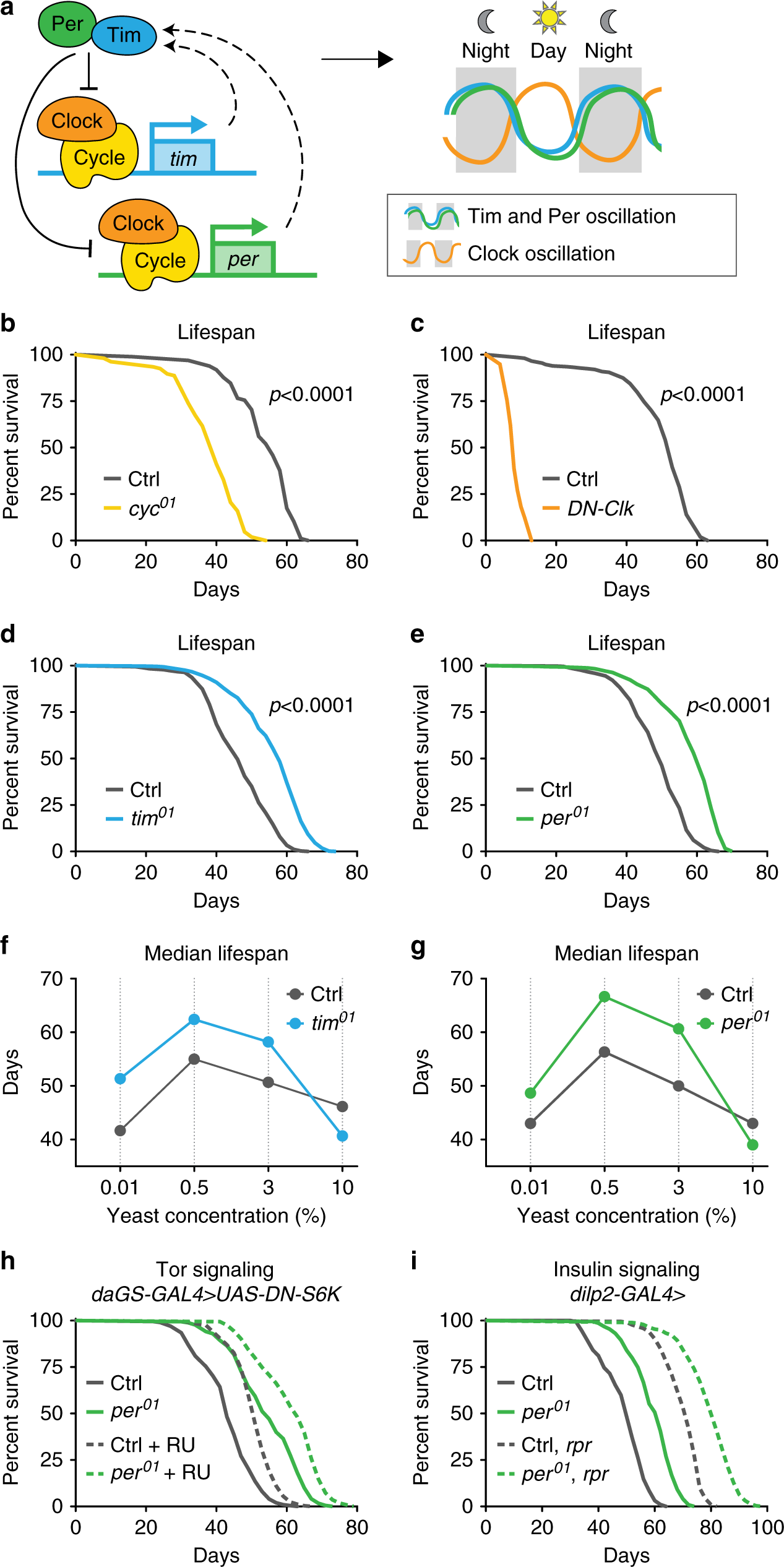
Fig. 1: Loss of the repressive arm of the transcriptional circadian clock extends male lifespan.
a Schematic of core molecular clock components and circadian transcriptional feedback loop. Relative to controls (gray), mutant males lacking Cyc function (yellow, b) or expressing dominant-negative clock (daGS > UAS-DN-Clock flies fed RU486, orange, c) show reduced lifespan (p < 0.0001 for each circadian mutant vs. control). In contrast, tim01 mutants (blue, d) and per01 mutants (green, e) live longer than controls (gray), even on diets with different concentrations of yeast extract (f, g), except for the highest yeast diet (p < 0.001 for each circadian mutant vs. control on each diet). h per01 mutants with daGS > UAS-DN-S6K fed either RU486 (dashed lines) or vehicle (solid lines) exhibited similar lifespan extension relative to controls containing daGS > UAS-DN-S6K. i per01 mutant males containing either the dilp2-GAL4 driver alone (solid lines) or dilp2-GAL4 > UAS-reaper and ablated for insulin-producing cells (dashed lines) exhibit similar lifespan extension relative to controls. See Supplementary Table 1 for n and p values for lifespan experiments, particularly multicurve comparisons; p values were obtained by log-rank analysis.
Here, we show that loss of the repressive arm of the core circadian clock extends male Drosophila lifespan. Loss of period also induces a highly active metabolic state characterized by increased mitochondrial uncoupling; this lifespan extension is due to upregulation of the endogenous mitochondrial uncoupling protein (UCP) UCP4C, specifically in the intestine. Loss of per or upregulation of UCP4C attenuates age-related decline in gut homeostasis. These genetic phenotypes, including longevity extension and improved gut homeostasis, are recapitulated by feeding Drosophila low doses of mitochondrial uncoupling drugs.
Results
Loss of tim or per extends lifespan in male Drosophila
To investigate how loss of specific circadian regulators influence aging, we examined the lifespans of four established arrhythmic Drosophila mutants (Fig. 1a): three genomic mutants, cycle (cyc01), period (per01), and timeless (tim01), and flies ubiquitously expressing a dominant-negative form of Clock (DN-Clock)21. Consistent with other reports3,4,5,6, functional disruption of the circadian transcriptional activators Cycle or Clock shortened the lifespan of male flies relative to controls (Fig. 1b, c). In contrast, functional disruption of the circadian transcriptional repressors Per and Tim significantly increased lifespan (15–20%) relative to controls; this appeared to be a male-specific effect (Fig. 1d, e). To confirm that lifespan extension was due to the loss of Per protein, we restored Per expression using the UAS–GAL4 system22. Expressing either of the two independent period transgenes using either the timeless–GAL4 driver or the ubiquitin–GAL4 driver in the per01 null background reverted per mutant lifespan to that of control animals (Supplementary Fig. 1A, B). Thus, loss of Per expression extends lifespan.
The classic method of lifespan extension is dietary restriction (DR). We showed previously that per01 and tim01 mutants are not diet-restricted—that is, these mutants eat more, not less, than controls (ref. 19; see also Fig. 2a). However, loss of Per and Tim might mimic physiological changes associated with DR. If so, DR should not further extend the lifespan of male per01 and tim01 mutants. In Drosophila, DR-mediated lifespan extension is accomplished by titration of protein (yeast extract (YE)). To test response to DR, we fed per01 and tim01 null mutants and controls four different concentrations of YE: 0.01% (low), 0.5% (DR), 3% (standard), and 10% (high). As we showed previously for female per01 and tim01 mutants10, male per01 and tim01 mutants exhibited DR-induced lifespan extensions similar to controls and lived longer than controls on most dietary protein concentrations (Fig. 1f, g). The lifespan of these circadian mutants was similar to that of control animals only at very high yeast concentrations (10%), which shortens lifespan. Thus, the extended longevity of per01 mutants appears to be independent of DR.
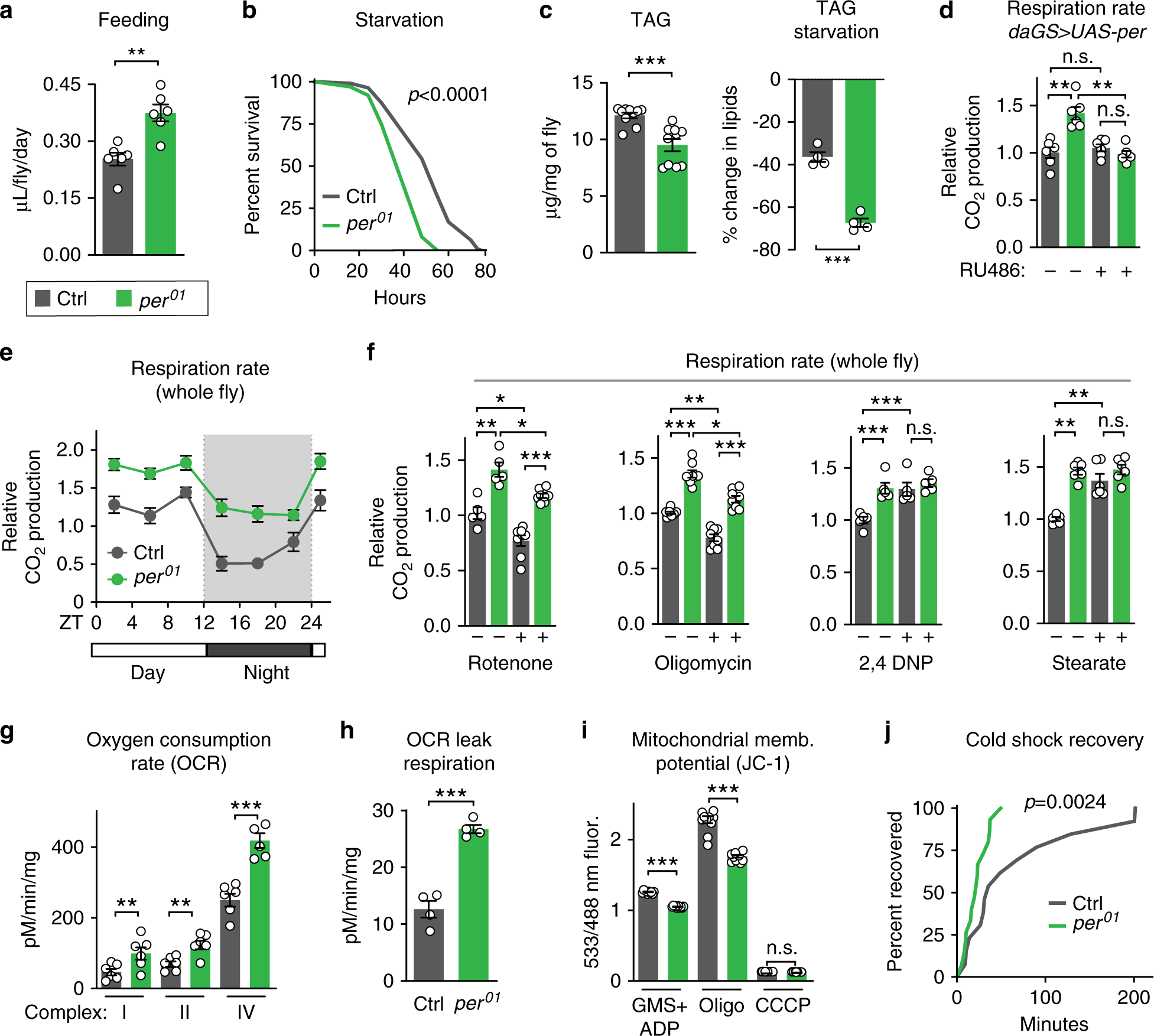
Fig. 2: period mutants exhibit high metabolic rate due to mitochondrial uncoupling.
Relative to controls (gray), per01 mutants (green) exhibited: a increased feeding rate (n = 6 vials of ten flies/condition, p < 0.01); b decreased survival upon starvation (n ≥ 99 flies per condition, p < 0.001); c lower baseline levels of lipids (left) and increased rate of lipid utilization after 24 h of starvation (right), as shown by quantification of triacylglyceride (TAG) levels (n ≥ 4 samples/condition, 5 flies/sample, both p < 0.0001); d increased respiration, which was reverted by ubiquitous overexpression of Per during adulthood (n = 6 groups of 10 flies per condition); e higher CO2 production over the circadian day (n = 6 groups of 10 flies/condition and timepoint); f higher respiration rates after 24 h of feeding with rotenone and oligomycin, but not with 2,4-DNP, or stearic acid (n ≥ 5 groups of 10 flies/condition); g increased oxygen consumption rate when stimulated through complexes I, II, and IV relative to controls (n = 4–6 oxygraph runs per condition); h increased leak respiration, using high-resolution respirometry on purified mitochondria (n = 5 oxygraph runs per condition, p < 0.001); i lower membrane potential, measured by JC-1 staining of purified mitochondria (n = 10 mitochondrial preps per condition, p < 0.001); and j faster recovery from cold shock (n = 26–30 flies per condition, p < 0.001). See Supplementary Information for n if not listed here; n.s.p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001; p values were obtained by unpaired two-tailed t-test (a, c, e, g–i), ANOVA followed by Tukey’s post-hoc test (d, f), and log-rank analysis (b, j); error bars represent SEM.
We next tested if per01 mutants exhibit canonical changes in DR-associated mechanisms of longevity, including: decreased insulin signaling, measured by decreased phosphorylation of Akt protein; decreased TOR signaling, measured by decreased phosphorylation of S6K; and increased autophagy, measured by increased lipidation of Atg8. Surprisingly, per01 males were atypical for these hallmarks of longevity and, at some points in the circadian cycle, exhibited the opposite phenotype as predicted for long-lived mutants (Supplementary Fig. 1C). We further used genetic manipulations to determine if the inhibition of TOR signaling or the inhibition of insulin-like signaling (ILS) is responsible for the longevity of per01 mutants. To test the inhibition of TOR signaling, we ubiquitously suppressed TORC1 signaling in adulthood by inducible (RU486-mediated) overexpression of a dominant-negative form of the downstream kinase S6K, which is known to extend lifespan23. The inhibition of TORC1 signaling extended the lifespans of both control animals and per01 mutants to a similar magnitude (Fig. 1h), suggesting that the longevity of per01 mutants is independent of TORC1 inhibition. Feeding RU486 to either controls or per01 mutants lacking the UAS transgene had no influence on lifespan (Supplementary Fig. 1D). To test the inhibition of ILS, we performed partial genetic ablation of insulin-producing cells in the fly brain using the proapoptotic gene reaper24. This extended the lifespans of both control animals and per01 mutants to a similar magnitude (Fig. 1i), suggesting that per01-associated lifespan extension is independent of insulin-like signaling inhibition. Together, these results suggest that the longevity phenotype of per01 males is not due to canonical longevity mechanisms but due to a different, independent pathway.
per mutants exhibit a high metabolic rate
As metabolism and lifespan are linked, we set out to investigate the metabolism of long-lived per01 mutants. As shown in our previous work19, per01 males exhibited hyperphagia (increased feeding), decreased starvation resistance, low levels of lipid storage, and increased starvation-induced lipid utilization relative to controls (Fig. 2a–c, Supplementary Fig. 2A). Because per01 mutants eat more but are leaner than controls, we tested for hyperactive metabolic rate by measuring CO2 production. Consistent with our previous characterization, per01 mutants produced more CO2 throughout the circadian cycle relative to controls, which was reverted by exogenous Per expression (Fig. 2d, e); respiration rate was not affected in RU486 feeding controls (Supplementary Fig. 2B). Respiration rate is significantly affected by the mitochondrial function of oxidative phosphorylation. To determine if increased oxidative phosphorylation caused this increased metabolic output, flies were fed sublethal doses of mitochondrial complex inhibitors: rotenone, which blocks complex I of the mitochondrial electron transport chain (ETC), or oligomycin, which blocks complex V, the F0F1 ATP synthase. While these compounds inhibited the CO2 output of both control and per01 animals to a similar degree, per01 flies still had higher respiration rates than controls (Fig. 2f). This suggests that their increased respiration is independent of mitochondrial ATP synthesis and instead implicates a different mitochondrial function, such as mitochondrial uncoupling.
Mitochondrial uncoupling increases respiration by dissipation of the proton gradient and uncoupling of oxidative phosphorylation from ATP synthesis, creating futile cycles of respiration and generating heat, as in mammalian brown fat. To test the effects of mitochondrial uncoupling on respiration, we fed per01 mutants and control flies two different mitochondrial uncoupling compounds: 2,4-dinitrophenol (2,4-DNP), a proton ionophore that dissipates the proton gradient; and stearic acid, which induces mitochondrial uncoupling by activating endogenous UCP activity. Treatment with either drug increased the respiration of control flies to levels similar to those of per01 mutants but did not increase the respiration of per01 mutants (Fig. 2f). These results suggest that the increased respiration of per01 mutants is due to increased mitochondrial uncoupling and that per01 mutants may already be maximally uncoupled within viable parameters. Higher doses of uncoupling drugs were lethal to both genotypes.
To directly test whether per01 mutants were mitochondrially uncoupled relative to controls, we assessed mitochondrial function in vitro. We purified mitochondria from per01 mutants and controls and measured O2 consumption. per01 mutant mitochondria exhibited increased O2 consumption when initiated through ETC complexes I, II, and IV (Fig. 2g). This increased O2 consumption was not due to increased mitochondrial ETC protein abundance or increased enzymatic activity (Supplementary Fig. 2C, D). Instead, per01 mitochondria exhibited two critical hallmarks of mitochondrial uncoupling relative to control mitochondria: increased leak respiration, measured by higher oxygen consumption after oligomycin treatment (Fig. 2h); and decreased membrane potential, or disrupted proton gradient, measured by the membrane potential dye sensor JC-1, both during steady-state ATP generation and after inhibition by oligomycin (Fig. 2i). Finally, to assess heat generation, another hallmark of mitochondrial uncoupling, we performed cold shock recovery assays on per01 mutants and control animals (Fig. 2j). After 1 h of cold shock at 4 °C, per01 mutants recovered significantly faster than controls, suggesting that per01 mutants may generate more heat than controls. Thus, loss of Per protein increases mitochondrial uncoupling.
Uncoupling protein UCP4C is required for per mutant longevity
To determine if this increased mitochondrial uncoupling is required for the lifespan extension of per01 mutants, we genetically manipulated the expression of endogenous proteins that cause mitochondrial uncoupling. Like many animals, Drosophila can undergo mitochondrial uncoupling by induction of UCPs, including UCP4A, B, and C25,26. Of these, we found that the expression of Ucp4B and Ucp4C is circadian-regulated in control flies and constitutively high in per01 mutants (Fig. 3a–c). To test the role of UCPs in the metabolic phenotype observed in per01 mutants, we first disrupted the expression of both UCP4B/C proteins using a mutant containing a piggyBac transposon in the intergenic region between these two closely linked Ucp4 genes (Supplementary Fig. 3A, B). Next, to determine if period nulls exhibit true mitochondrial uncoupling via classic mitochondrial UCPs, we tested stimulation of mitochondrial respiration in the presence of palmitate and reversal by the UCP inhibitor guanosine nucleotide (GTP) suppression of respiration. Palmitate and GTP are respectively known to stimulate and suppress classic mitochondrial UCPs27,28,29. Indeed, per01 flies showed significantly increased respiration in the presence of palmitate, which was reversed by the addition of GTP (Supplementary Fig. 3C). per01 flies with disrupted UCP4B/C expression showed no response to palmitate or GTP indicating that the uncoupled respiration is due to UCP gene function. In addition, disruption of UCP4B/C in per01 flies reverted other hallmarks of uncoupling: mitochondrial leak respiration (Fig. 3d); mitochondrial membrane potential (Fig. 3e); mitochondrial oxygen consumption rate (Supplementary Fig. 3D); and whole-animal cold shock recovery rates (Fig. 3f). Disruption of the UCP4B/C expression not only reverted mitochondrial uncoupling but also reverted per01 lifespan to that of controls, suggesting that mitochondrial uncoupling causes lifespan extension (Fig. 3g). To inhibit UCPs by an orthogonal mechanism, we performed RNAi-mediated knockdown of UCP4A, B, and C in adulthood throughout the whole body in both per01 mutants and controls. Consistent with our mutant analysis, knockdown of UCP4B or UCP4C, but not UCP4A, reverted per01 lifespan to that of control animals (Supplementary Fig. 3E–G). Thus, the Ucp4B/C expression is necessary for the longevity and metabolic phenotypes of per01 mutants.
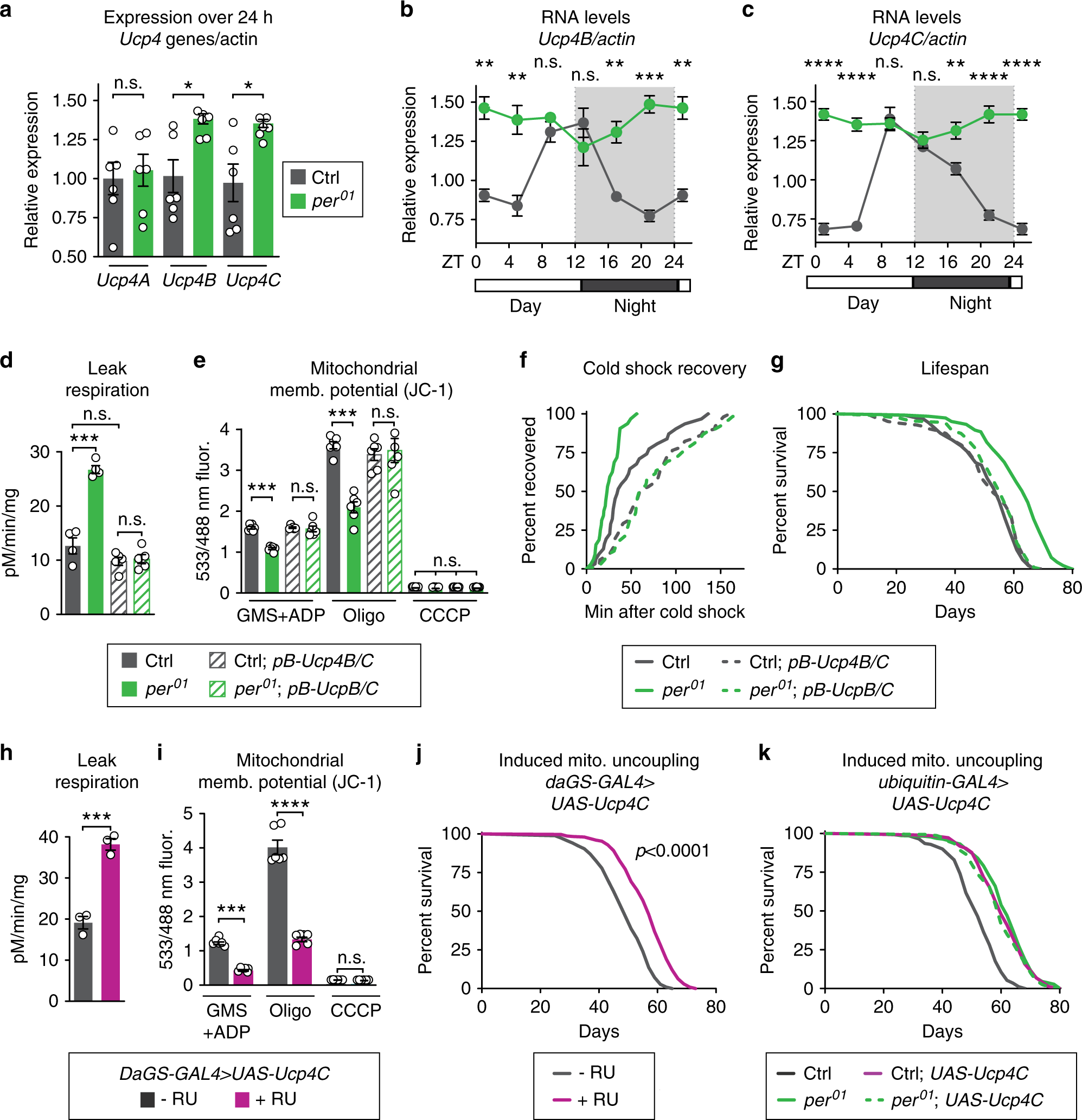
Fig. 3: UCP4C is necessary for period mutant lifespan and sufficient to extend wild-type lifespan.
Relative to controls (gray), per01 mutants (green) exhibited: a higher expression of Ucp4B and Ucp4C but not Ucp4A; and constitutively high expression of b Ucp4B and c Ucp4C, both of which are circadian-regulated in controls. Relative to controls (gray), per01 mutants (green) also exhibited the following phenotypes, which were reverted by suppression of Ucp4B/C expression, comparing flies with (dashed lines) or without (solid lines) piggyback mutation of Ucp4B/C: d increased leak respiration by purified mitochondria; e decreased mitochondrial membrane potential; f faster cold shock recovery; and g increased lifespan. Relative to vehicle-fed controls (gray), daGS > UAS-Ucp4C flies fed RU486 to induce constitutive UCP4C overexpression (magenta) exhibited: h higher leak respiration (p < 0.001); i lower mitochondrial membrane potential (p < 0.001); and j increased lifespan (p < 0.0001). k Ubiquitous overexpression of UCP4C in otherwise wild-type flies was sufficient to extend lifespan (gray, dashed) relative to driver-only controls (gray, solid). In per01 mutants (green, solid), overexpression of UCP4C did not further extend the lifespan of per01 mutants (green, dashed). See Supplementary Table 1 for n and statistical analysis of lifespans, particularly multicurve comparisons; n.s.p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001; p values were obtained by unpaired two-tailed t-test (a, h, i), ANOVA followed by Tukey’s post-hoc test (d, e), and log-rank analysis (f, g, j, k); error bars represent SEM.
Finally, to determine if increased UCP expression is sufficient to extend the lifespan of WT animals, we ubiquitously overexpressed UCP4C at low levels during adulthood using conditional GeneSwitch drivers induced by the drug RU486. Flies overexpressing UCP4C showed mitochondrial uncoupling phenotypes very similar to per01 mutants; increased mitochondrial leak respiration (Fig. 3h), decreased mitochondrial membrane potential (Fig. 3i), increased CO2 production (Supplementary Fig. 4A), and faster cold shock recovery (Supplementary Fig. 4B). Most importantly, the constitutive expression of UCP4C extended the lifespan of otherwise wild-type flies to the same extent as per01 mutants, with no effect on per01 mutants (Fig. 3j–k). Feeding these low doses of RU486 alone had no effect on either metabolic or lifespan phenotypes when flies lacked the UAS transgene (Supplementary Fig. 3C–G). These results suggest that UCP4C functions in the same pathway as Per to extend per01 mutant lifespan and that increased expression of the mitochondrial uncoupling protein UCP4C extends lifespan. Taken together, our data point to mitochondrial uncoupling as the major circadian-regulated physiology directly responsible for the extended lifespan of per01 mutants.
.../...
Edited by Engadin, 06 May 2020 - 05:58 PM.















































