































.
Abstract
DNA methylation has fundamental roles in gene programming and aging that may help predict mortality. However, no large-scale study has investigated whether site-specific DNA methylation predicts all-cause mortality. We used the Illumina-HumanMethylation450-BeadChip to identify blood DNA methylation sites associated with all-cause mortality for 12, 300 participants in 12 Cohorts of the Heart and Aging Research in Genetic Epidemiology (CHARGE) Consortium. Over an average 10-year follow-up, there were 2,561 deaths across the cohorts. Nine sites mapping to three intergenic and six gene-specific regions were associated with mortality (P < 9.3x10-7) independently of age and other mortality predictors. Six sites (cg14866069, cg23666362, cg20045320, cg07839457, cg07677157, cg09615688)—mapping respectively to BMPR1B, MIR1973, IFITM3, NLRC5, and two intergenic regions—were associated with reduced mortality risk. The remaining three sites (cg17086398, cg12619262, cg18424841)—mapping respectively to SERINC2, CHST12, and an intergenic region—were associated with increased mortality risk. DNA methylation at each site predicted 5%–15% of all deaths. We also assessed the causal association of those sites to age-related chronic diseases by using Mendelian randomization, identifying weak causal relationship between cg18424841 and cg09615688 with coronary heart disease. Of the nine sites, three (cg20045320, cg07839457, cg07677157) were associated with lower incidence of heart disease risk and two (cg20045320, cg07839457) with smoking and inflammation in prior CHARGE analyses. Methylation of cg20045320, cg07839457, and cg17086398 was associated with decreased expression of nearby genes (IFITM3, IRF, NLRC5, MT1, MT2, MARCKSL1) linked to immune responses and cardiometabolic diseases. These sites may serve as useful clinical tools for mortality risk assessment and preventative care.
Introduction
The human epigenome contains DNA methylation marks that progressively change as we age. DNA methylation can influence gene expression and manifests in response to both environmental and hereditary factors [1, 2]. Biological age estimations, constructed from DNA methylation marks and referred to as “epigenetic aging clocks”, have been associated with environmental exposures, morbidities, and mortality [9–13]. As these clocks were designed to track chronological age, not to predict mortality, further study is necessary to fully elucidate indicators of all-cause mortality. To date, no large-scale analysis has been conducted to identify variations in DNA methylation at individual 5’-cytosine-phosphate-guanosine-3’ (CpG) sites associated with future mortality risk. Here, we present an epigenome-wide methylation analysis of 12,300 participants and 2, 561 (21%) deaths from 12 American and European cohorts to determine whether site-specific DNA methylation predicts all-cause mortality, independent of age, lifestyle factors, and clinical predictors of mortality including comorbidities. We also assessed the causal relationship of identified sites with age-related chronic diseases using Mendelian randomization approaches, and we related the sites to epigenetic aging clocks and a mortality risk score, an epigenetic indicator of mortality previously created and validated with DNA methylation arrays in two European cohorts.
Results
Cohorts
Across studies in the Cohorts of the Heart and Aging Research in Genetic Epidemiology (CHARGE) Consortium, mortality rates ranged from 3%–70% of all participants, and the average time to death or censoring ranged from 4.4–16.6 years (Supplementary Table 1). Each study conducted epigenome-wide mortality analyses, adjusting for two sets of harmonized risk factors and confounders, and shared results for meta-analysis (Figure 1).
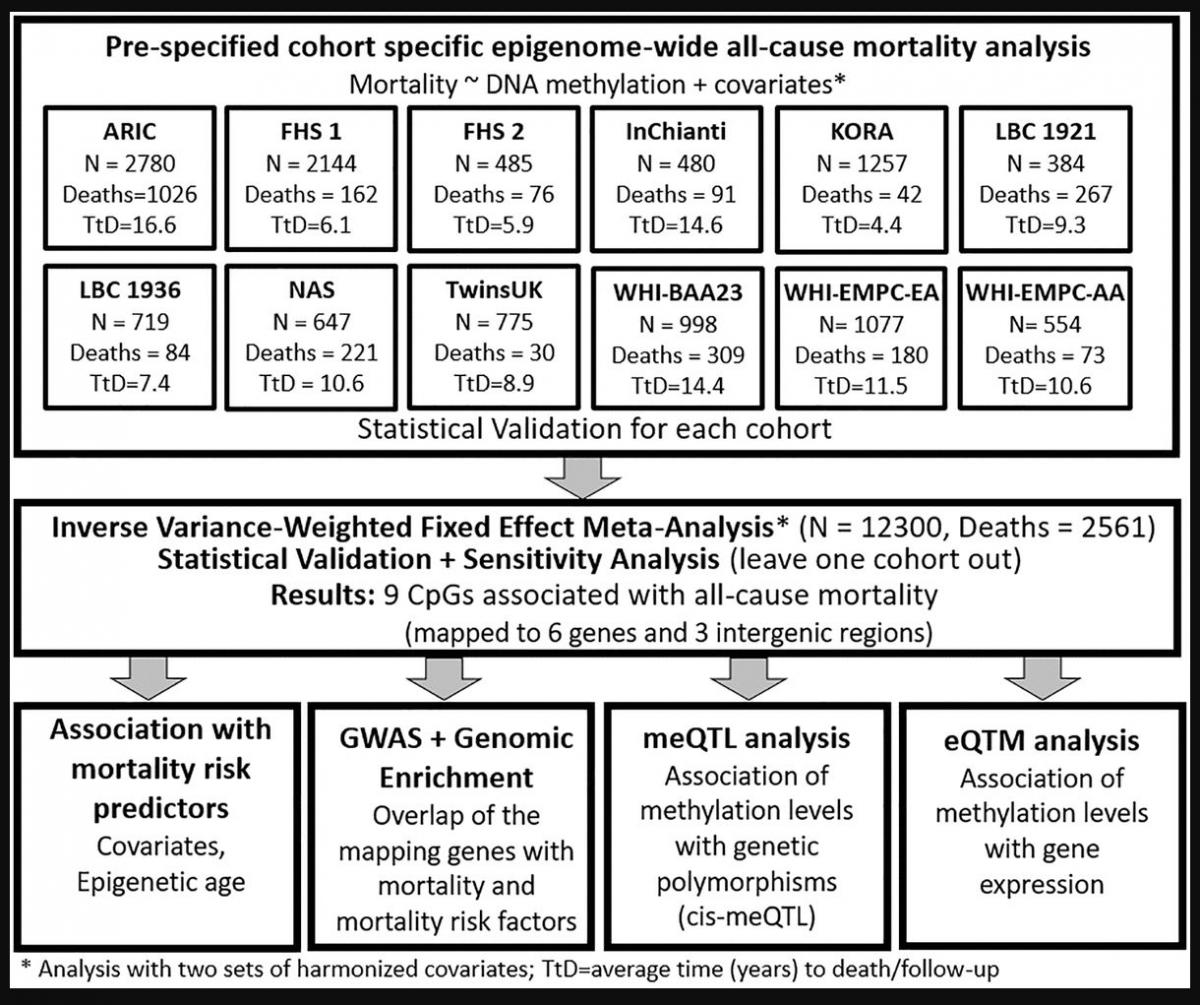
Figure 1. Workflow of the study.
Meta-analysis
Inverse variance-weighted fixed-effects meta-analysis of 426, 724 CpGs identified 51 Bonferroni-significant and 257 FDR-significant (P < 3.03x10-5) CpGs in a basic model adjusting for age, sex, technical covariates, and white blood cell proportions (Figure 2A and Supplementary Table 2). We also identified three Bonferroni-significant and nine FDR-significant (P < 9.3x10-7) CpGs in a fully-adjusted model also adjusting for education, smoking status, pack-years smoked, body mass index, recreational physical activity, alcohol consumption, hypertension, diabetes, and history of cancer and coronary heart disease (Figures 2B, 3A and Supplementary Table 3). For 188 (73%) basic-adjusted FDR-significant CpGs and six (67%) fully-adjusted CpGs, higher blood DNA methylation was associated with lower all-cause mortality (Figure 2 and Supplementary Tables 2, 3).
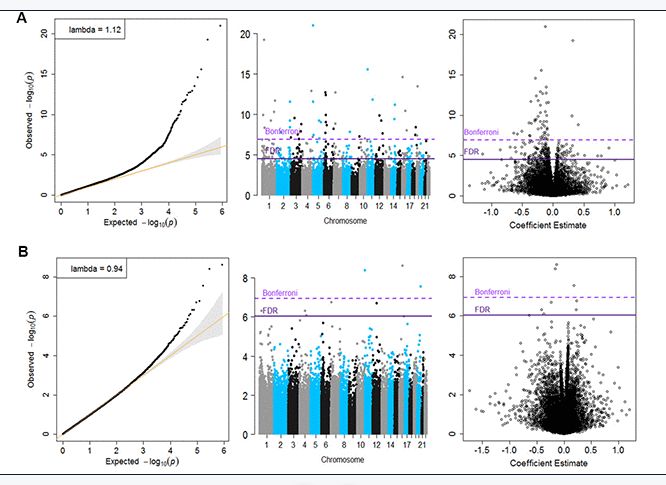
Figure 2. Quantile-Quantile plots, Manhattan and Volcano for the basic model (Panel A) and for the fully adjusted model (Panel B).
All nine fully-adjusted FDR-significant CpGs had similar magnitude of associations with mortality in the basic model, although only five were also FDR-significant in the basic model (Figure 3B). Hazard ratios (HRs) of the nine fully-adjusted FDR-significant CpGs ranged 0.53–1.26 per 10% increase in DNA methylation levels, where 1 represents 100% methylation (Supplementary Table 4). Six sites (cg14866069, cg23666362, cg20045320, cg07839457, cg07677157, cg09615688) were associated with reduced mortality risk, while the remaining three sites (cg17086398, cg12619262, cg18424841) were associated with increased mortality risk (Figure 3A and Supplementary Tables 3, 4). Three fully-adjusted CpGs (cg07677157, cg09615688, cg18424841) were in intergenic regions; the remaining six (cg17086398, cg14866069, cg23666362, cg12619262, cg20045320, cg07839457) were within 10,000 bp of a gene, with two CpGs (cg07839457, cg23666362) mapped respectively to nucleotide-binding oligomerization domain-like receptor caspase recruitment domain containing 5 (NLRC5) and microRNA 1973 (MIR1973) within 1,500 bp of transcription start sites, and one (cg17086398) in the serine incorporator 2 (SERINC2) gene body (Supplementary Table 3).
.../...
.














