.
F U L L T E X T S O U R C E : Cell
Abstract
Age-related tissue alterations have been associated with a decline in stem cell number and function. Although increased cell-to-cell variability in transcription or epigenetic marks has been proposed to be a major hallmark of ageing, little is known about the molecular diversity of stem cells during ageing. Here we present a single cell multi-omics study of mouse muscle stem cells, combining single-cell transcriptome and DNA methylome profiling. Aged cells show a global increase of uncoordinated transcriptional heterogeneity biased towards genes regulating cell-niche interactions. We find context-dependent alterations of DNA methylation in aged stem cells. Importantly, promoters with increased methylation heterogeneity are associated with increased transcriptional heterogeneity of the genes they drive. These results indicate that epigenetic drift, by accumulation of stochastic DNA methylation changes in promoters, is associated with the degradation of coherent transcriptional networks during stem cell ageing. Furthermore, our observations also shed light on the mechanisms underlying the DNA methylation clock.
Introduction
Epigenetic alterations have been proposed to be a major cause of age-related decline in tissue function1. Changes in DNA methylation are well correlated with ageing, and methylation of specific loci has been used to predict age in a large number of tissues1,2. Epigenetic-based age predictive models, also referred to as epigenetic clocks, have been widely applied in the last few years and are believed to reflect chronological aging3. Despite their accuracy, the mechanisms underlying these models are largely unknown2,3.
Notwithstanding the close associations with age, age-related methylation changes are poorly correlated with transcriptional variation, presumably because the changes are generally small and may not occur homogeneously in all cells2, a phenomenon also known as epigenetic drift. Although epigenetic drift has long been hypothesised to be an important hallmark of ageing4, this proposal has been challenging to test because of technical constraints. However, powerful combined single cell methods5,6 are now available, and epigenetic changes during ageing together with their functional consequences can now be read out in single cells7.
Degenerative changes in tissue-specific stem cells have been proposed to be a major cause of age-related decline in tissue function8. While several reports indicate a loss of clonal diversity during early life stages9,10,11, little is known about how cell-to-cell variability at the molecular level is involved in stem cell ageing. Here, we performed parallel single-cell DNA methylation and transcriptome sequencing (scM&T-seq) on the same cell6 to investigate how ageing affects transcriptional and epigenetic heterogeneity of tissue-specific stem cells, using mouse muscle stem cells as a model. Muscle stem cells express the transcription factor Pax712 and are largely quiescent in adult muscles. They activate upon injury to differentiate and fuse to form new fibres, or self-renew to reconstitute the stem cell pool12. Age-associated muscle defects have been attributed to a decrease in stem cell number together with impaired regenerative potential13. In addition, clonal lineage-tracing of mouse muscle stem cells showed that population diversity is unaltered during homoeostatic ageing14.
Here, by combined single-cell transcriptome and DNA methylome profiling in muscle stem cells, we show a global increase of uncoordinated transcriptional heterogeneity and context-dependent alterations of DNA methylation with age. Notably, old cells that change the most with age reveal alterations in the transcription of genes regulating cell-niche interactions. These findings show linked increases in heterogeneity between the epigenome and the transcriptome, with consequent degradation of coherent transcriptional networks and stem cell functional decline during ageing.
Results
Transcriptomic profiling of young and old muscle stem cells
Muscle stem cells with high expression of Pax7 were shown to be in a deep quiescent, or dormant, state15,16. To investigate the molecular effects of ageing in a defined population that is less poised to enter the cell cycle, we isolated single muscle stem cells by fluorescence-activated cell sorting (FACS) from young (1.5 months) and old (26 months) Tg:Pax7-nGFP mice17 and selected those with high levels of GFP, to which we applied scM&T-seq (Fig. 1a). Importantly, this approach allows us to study variability while minimising key confounder factors such as differences in cell cycle or differentiation states between ages.
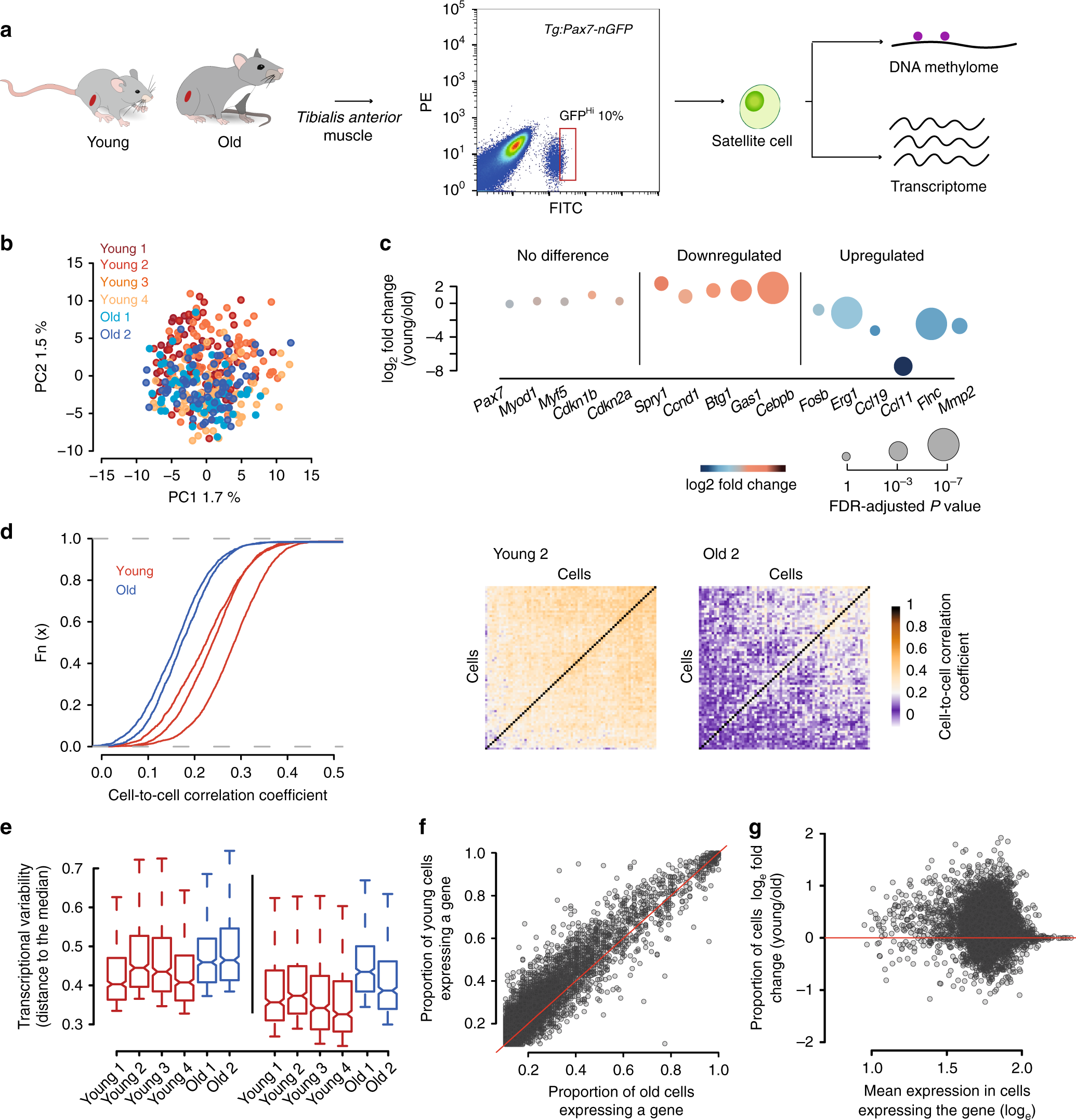
Aged muscle stem cells have increased cell-to-cell transcriptional variability. a Experimental scheme. Single cells were isolated from Tg:Pax7-nGFP young and old mice and subjected to parallel single-cell methylation and RNA sequencing. b Principal Component Analysis (PCA) of a total of 377 cells from young (n = 4) and old (n = 2) individuals. c Selected markers and differentially expressed genes between young and old cells. Source data are provided in Supplementary Data 1. d Cumulative distribution of cell-to-cell Spearman correlation values per individual (left) showing that transcriptional heterogeneity dramatically increases with age. Heatmap showing cell-to-cell Spearman correlation values from a young and an old mouse (right). e Distance to the median of the top 500 most variable genes among all genes (left) and of the top 500 most variable genes among the 5127 common genes expressed in the six individuals (right). For all boxplots, the box represents the interquartile range and the horizontal line in the box represents the median (n = 500). f Frequency of gene expression in young and old cells. Note that the proportion of cells expressing a given gene is reduced in old cells. g Independence between frequency of gene expression differences and gene expression level
After quality control and filtering, a total of 377 transcriptomes from four young and two old mice were analysed. Young (n = 253) and old (n = 124) cells from different individuals clustered together, respectively, indicating no global differences with age and absence of sequencing-related batch effects (Fig. 1b). We also assigned a cell cycle stage18 to each cell and observed that, with the exception of one cell from sample Young 2, cells were unlikely to be cycling at the time of isolation, and showed no differences between ages (Supplementary Fig. 1). Measures of cell cycle entry through BrdU uptake in vivo are also in agreement with this observation, supporting the deep quiescent state of these cells (Supplementary Fig. 2). Furthermore, we did not observe significant differences in the levels of Pax7, the myogenic factors Myod and Myf5 and the cell cycle inhibitor Cdkn1b (p27), nor of senescent markers such as Cdkn2a (p16INK4a/p14arf), suggesting that some molecular signatures are conserved between the analysed cell populations (Fig. 1c). Nevertheless, 940 genes were differentially expressed between young and old individuals (SCDE, FDR P < 0.05, Supplementary Data 1, Supplementary Fig. 3), with small differences in some cases; Spry1, which is a key factor for maintaining quiescence19, and the cell cycle regulators Ccnd1 (Cyclin D1), Btg1 and Gas1 were down-regulated, while ageing markers such as the chemokine genes Ccl11 and Ccl19 were upregulated16(Fig. 1c). Furthermore, we identified significant alterations in expression of genes not previously reported to change in expression with age, such as the early activation markers Fosb and Egr120 and the metalloproteinase Mmp2 (Fig. 1c).
Increased transcriptional variability with age
To investigate if ageing affects transcriptional heterogeneity of the stem cell pool, we calculated pairwise correlation coefficients between cells within each individual (see “Methods”) and observed that old individuals showed consistently lower correlation (1.3 mean-fold decrease, Mann–Whitney–Wilcoxon test; P < 2.2e-16, Fig. 1d), indicating a remarkably lower degree of similarity between cells and no obvious population substructure. Similar results were observed when performing the analysis on cohorts of 10 cells from the same individual where the mean correlation in young individuals was always higher than in old ones (P < 0.001) (Supplementary Fig. 4).
We also computed an expression-level normalised measure of gene expression heterogeneity (named distance to the median)21, which proved to be higher in old individuals (Mann–Whitney–Wilcoxon test; P < 2.2e-16, Fig. 1e), revealing a striking global increase of uncoordinated transcriptional variability with age. Notably, the proportion of cells expressing a given gene (frequency of gene expression) was reduced with age (Mann–Whitney–Wilcoxon test; P < 2.2e-16, Fig. 1f), even for genes that did not significantly change mean expression levels (SCDE, FDR P > 0.05, Fig. 1f). Importantly, we observed that this was independent of gene expression levels and not restricted to lowly expressed genes (Fig. 1g), suggesting that this global feature is not due to technical effects.
Genes that displayed increased expression variability with age (expression frequency difference > 15%) were enriched in extracellular matrix processes and include several collagen genes (Col4a2, Col5a3, Col4a1) and other extracellular matrix-related genes such as Dag1, Sparc, Cdh15, Lamc3 or Itgb1 (Fig. 2a, Supplementary Figs. 5 and 6). Interestingly, muscle stem cells without Itgb1 (β1-integrin) cannot maintain quiescence, and its experimental activation improves ageing-related decline in muscle regeneration22. Similarly, reduction of N-cadherin and M-cadherin (Cdh15) leads to a break of quiescence of muscle stem cells23. Notably, none of the above-mentioned genes were shown to change in expression level during the isolation procedure of muscle stem cells24.
.../...
F O R T H E R E S T O F T H E S T U D Y , P L E A S E V I S I T T H E S O U R C E .
.















































