































.
F U L L T E X T S O U R C E : Genome Biology
Abstract
Background
Brain aging is a complex process that depends on the precise regulation of multiple brain regions; however, the underlying molecular mechanisms behind this process remain to be clarified in non-human primates.
Results
Here, we explore non-human primate brain aging using 547 transcriptomes originating from 44 brain areas in rhesus macaques (Macaca mulatta). We show that expression connectivity between pairs of cerebral cortex areas as well as expression symmetry between the left and right hemispheres both decrease after aging. Although the aging mechanisms across different brain areas are largely convergent, changes in gene expression and alternative splicing vary at diverse genes, reinforcing the complex multifactorial basis of aging. Through gene co-expression network analysis, we identify nine modules that exhibit gain of connectivity in the aged brain and uncovered a hub gene, PGLS, underlying brain aging. We further confirm the functional significance of PGLS in mice at the gene transcription, molecular, and behavioral levels.
Conclusions
Taken together, our study provides comprehensive transcriptomes on multiple brain regions in non-human primates and provides novel insights into the molecular mechanism of healthy brain aging.
Background
Aging, an intricate and irreversible process, varies significantly at the individual level, depending on a combination of genetic and environmental factors an individual experiences throughout a lifetime [1,2,3]. Aging is associated with cognitive decline and memory loss and has been implicated in many neurodegenerative disorders [4,5,6,7,8], thereby posing a major threat to global health. Despite its ubiquity and importance, aging-related alterations have mainly been observed by histology and ethology [9,10,11], with the underlying molecular mechanisms remaining elusive.
Aging processes are reliant on precise spatiotemporal regulation of the transcriptome, and changes in gene expression have been studied widely in brain aging [12,13,14,15]. However, an increasing body of persuasive evidence suggests that aging-related changes depend on the coordination of diversified transcriptional regulation rather than gene expression only [16,17,18,19,20]. One essential mechanism for increasing the spatiotemporal complexity of the transcriptome is alternative splicing, which generates multiple mRNA transcripts from a single gene and affects up to 95% of human multi-exon genes [21]. Moreover, the brain expresses more alternative splicing transcripts than any other tissue [21,22,23], and dysregulation of alternative splicing may affect healthy brain aging [16].
Currently, emerging evidence from human and animal models suggests that brain aging is regulated by the interaction of multiple brain regions [24], which must work together as a network to control this complex physiological process. Nevertheless, previous research has been limited to only a few brain regions (frontal/prefrontal cortex) [25,26,27]. Currently, the lack of a genome-wide transcriptional landscape of multiple brain regions limits our understanding of how spatiotemporal orchestration of the transcriptome regulates the process of brain aging.
The advent of high-throughput RNA-sequencing (RNA-seq) has allowed for a much more comprehensive exploration of brain aging. In this study, we sequenced the transcriptomes of 44 brain areas from 4 young and 3 aged rhesus macaques (Macaca mulatta) (Fig. 1), which are close non-human primate (NHP) relatives of humans (diverging 25 million years ago) [28, 29], to survey transcriptional profile alterations during aging. Through multifaceted analyses of RNA-seq data and integration of gene expression and alternative splicing (Fig. 1), we provide several novel insights into the molecular underpinnings of brain aging. In addition, we discovered a novel hub gene, PGLS, underlying brain aging and confirmed its function at the molecular and phenotypic level in mice.
Fig.1
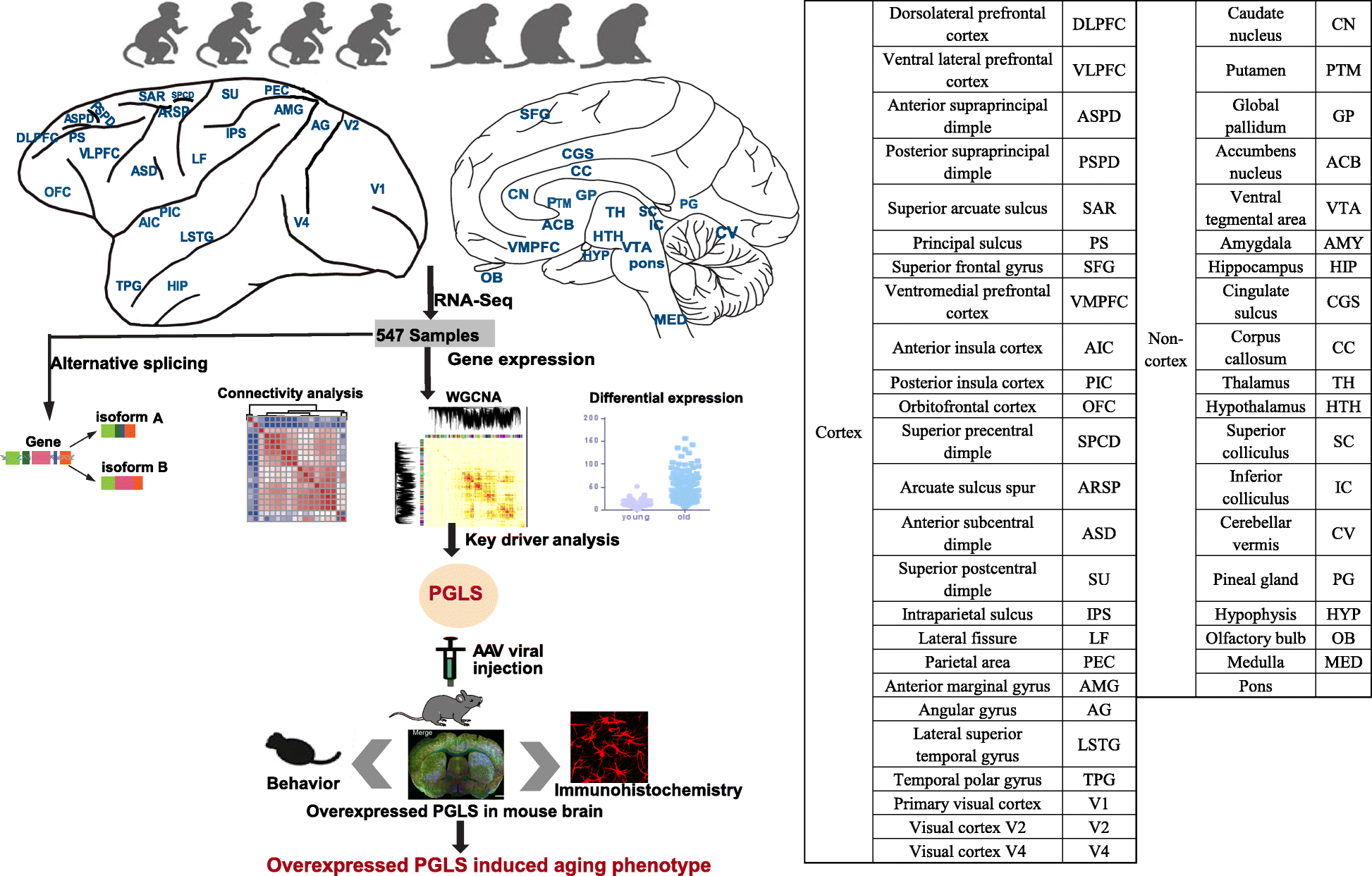
Schematic view of this study. We used 4 young and 3 aged macaques across 44 brain regions to study aging mechanism in NHPs through multifaceted analyses (connectivity analysis, differentially expressed gene analysis, alternative splicing analysis, and network analysis). We further confirmed the role of PGLS underlying brain aging in mice. The table on the right shows the ontology and nomenclature of analyzed brain regions.
Results
Transcriptome profiling across multiple brain areas in rhesus macaques
To investigate the dynamic expression patterns associated with brain aging, we used deep RNA-seq to profile transcriptomes from 590 post-mortem samples isolated from 44 brain areas across the left and right hemispheres in 4 young (5, 6, 6, and 6 years old) and 3 aged (16, 17, and 24 years old) rhesus macaques (Fig. 1). Rhesus macaques reach sexual maturity at 3–4 years and have a typical lifespan of 20 to 30 years in captivity [30]. After rigorous quality control (see the “Methods” section; Additional file 1: Figure S1), 547 samples remained for downstream analyses. RNA-seq data were then normalized, and genes with low expression values were removed to reduce the influence of technical noise. Consequently, 15,531 (61.8%) out of 25,111 genes were detected as having expression signals (an expressed gene was identified as having least 10 fragments in 80% of samples). There were no significant differences in the RNA integrity numbers (Mann-Whitney U (MWU) test, p = 0.723, N = 547) or post-mortem intervals (MWU test, p = 1, N = 547) between samples originating from young and aged groups (Additional file 2: Table S1). Sex, hemisphere, brain region, and individual did not explain a significant amount of expression variation. In contrast, most of the variation in gene expression could be attributed to age (p = 0.006; Additional file 2: Table S2), suggesting that age contributes more to global differences in gene expression than any of the other tested variables.
After accounting for the effects of many known biological and technical confounding factors, we performed principal component analysis (PCA) on gene expression in the 547 samples and found that the cortex and non-cortex clearly clustered into 2 separate groups (Additional file 1: Figure S2a). Hierarchical clustering analysis based on inter-array correlation also showed distinct clustering of these two groups (Additional file 1: Figure S2b). Thus, for the following, we studied the cortex and non-cortex in the downstream analyses separately.
Attenuation of expression connectivity during brain aging in NHPs
To assess the changing tendency of transcriptional connectivity among macaque brain regions during aging, we determined the expression correlation between any two brain regions in young and aged groups, respectively. By comparing the correlation matrices at different ages, we found that inter-areal correlations within the cortex decreased after aging (Pearson’s correlation: p = 2.00e−09, MWU test), with a less dramatic shift seen in the non-cortex (Pearson’s correlation: p = 0.075, MWU test) (Fig. 2a). Moreover, pairwise comparisons of gene expression across all regions of the cortex showed an increase in the number of differentially expressed genes (DEGs) between paired regions during aging (p = 0.009, MWU test; Additional file 1: Figure S3a), but no significant change was seen in the non-cortex (p = 0.2, MWU test; Additional file 1: Figure S3b). Our results suggest that attenuation of expression connectivity occurs in the cerebral cortex during aging. To further validate this observation, we repeated the correlation analysis using another public age-matched human transcriptome dataset (12–20 to over 60 years old; sampled brain areas can be seen in Additional file 2: Table S3) [31]. Similarly, the expression connectivity among human brain areas was substantially decreased in the cortex (p = 1.12e−12, MWU test) but unchanged in the non-cortex (p = 0.35, MWU test) after brain aging (Additional file 1: Figure S4), supporting the robustness of our results and indicating conserved and consistent changes in expression connectivity during brain aging in primates.
Fig. 2
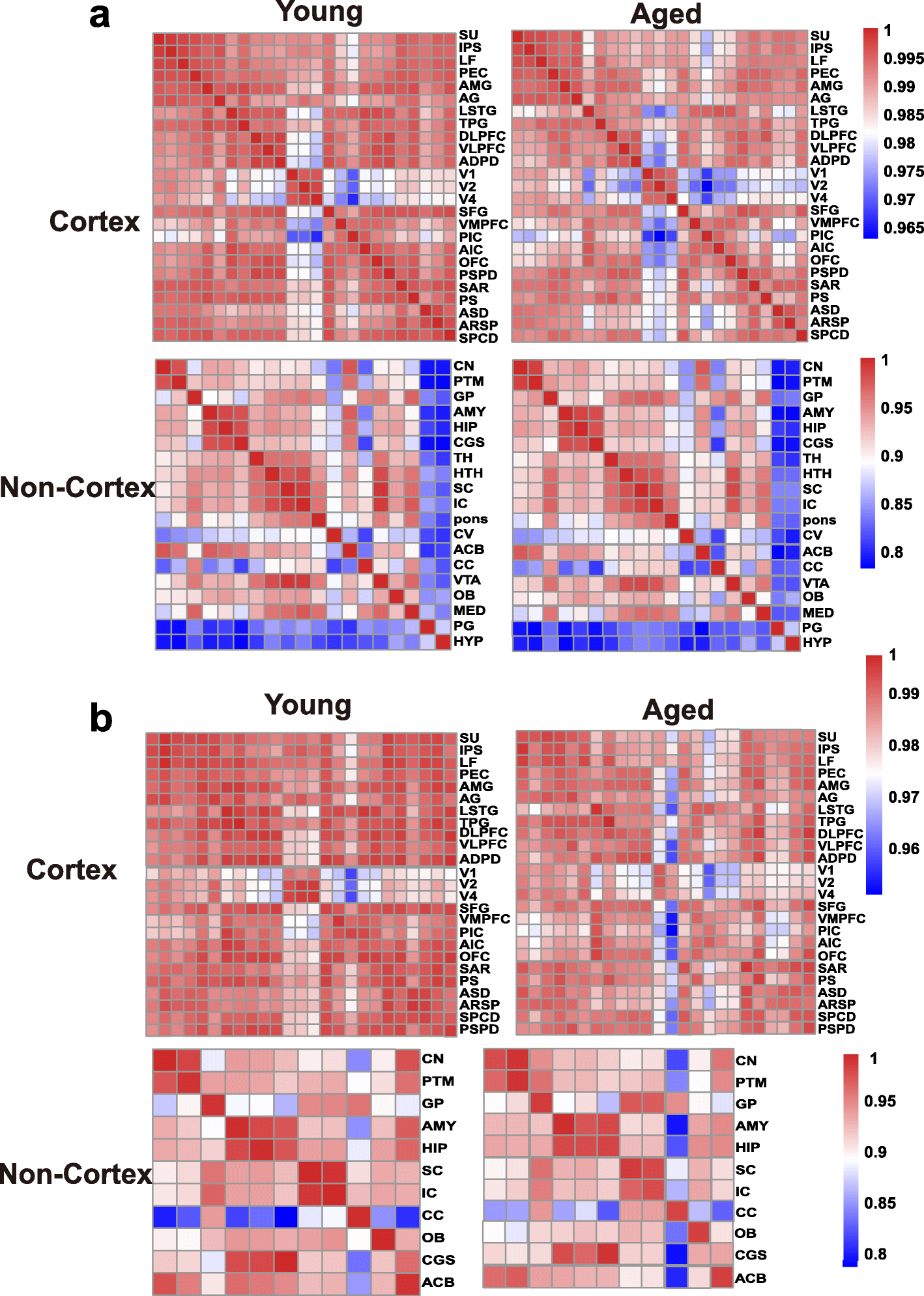
Expression connectivity between pairs of cerebral cortex areas and expression symmetry between the left and right hemispheres decrease after aging. a Heat map matrix of pairwise Pearson correlations between cortex regions (top) and between non-cortex areas (bottom) in young and aged macaques. b Heatmap matrix of pairwise Pearson correlations between the left and right hemispheres in cortex (top) and non-cortex (bottom) regions in young and aged macaques (columns represent brain areas across the left hemisphere; rows represent brain areas across the right hemisphere).
Additionally, by performing unsupervised hierarchical clustering on the multiple brain areas based on the gene expression signals in the young and aged groups (Additional file 1: Figure S5), we found that the relative relationship between some brain regions was altered during brain aging. For example, the ventromedial prefrontal cortex (VMPFC) clustered closely with the posterior insula cortex (PIC) in young macaques but shifted towards the anterior insula cortex (AIC) and lateral superior temporal gyrus (LSTG) in the aged group, suggesting a dynamic reorganization of transcriptional patterns between brain areas during aging.
The brain hemispheres are known to be anatomically and functionally asymmetric [32, 33]. Thus, to explore changes in expression connectivity between the left and right hemispheres during aging, we determined the correlation matrices of pairwise comparisons between the hemispheres at different ages. Results showed that in the cortex, the correlation coefficient in the young group was significantly higher than that in the aged group (p = 0.00011, MWU test), but unchanged in the non-cortex (p = 0.7541), suggesting a decreased tendency of transcriptome connectivity between the left and right hemispheres in the cortex during aging (Fig. 2b).
Transcriptional changes across multiple brain areas during NHP aging
We next investigated the broad patterns of aging-related transcriptome changes in each brain area by integration of gene expression (p < 0.05, fold change [FC] > 1.5) and alternative splicing (p < 0.01). Results showed a positive correlation between the number of DEGs and genes with differential exon usage genes (DEUs) across brain regions (Pearson’s r2 = 0.39, p = 0.018; Additional file 1: Figure S6). In addition, changes in gene expression and alternative splicing were widespread in all brain regions, although the changes were not uniform (Fig. 3a). Focusing on the 37 brain regions with similar sample sizes, the visual cortex V4 area was the most prioritized region exhibiting changes in gene expression (Fig. 3a). In terms of alternative splicing, the putamen (PTM) was the most pronounced region showing aging-related changes (Fig. 3a). Although the putamen plays an important role in cognitive ability [34,35,36], few studies have focused on its role in brain aging.
.../...
F O R T H E R E S T O F T H E S T U D Y, P L E A S E V I S I T T H E S O U R C E .
.













