































.
Abstract
Despite a growing number of studies on longevity in Drosophila, genetic factors influencing lifespan are still poorly understood. In this paper we propose a conceptually new approach for the identification of novel longevity-associated genes and potential target genes for SNPs in non-coding regions by utilizing the knowledge of co-location of various loci, governed by the three-dimensional architecture of the Drosophila genome. Firstly, we created networks between genes/genomic regions harboring SNPs deemed to be significant in two longevity GWAS summary statistics datasets using intra- and inter-chromosomal interaction frequencies (Hi-C data) as a measure of co-location. These networks were further extended to include regions strongly interacting with previously selected regions. Using various network measures, literature search and additional bioinformatics resources, we investigated the plausibility of genes found to have genuine association with longevity. Several of the newly identified genes were common between the two GWAS datasets and these possessed human orthologs. We also found that the proportion of non-coding SNPs in borders between topologically associated domains is significantly higher than expected by chance. Assuming co-location, we investigated potential target genes for non-coding SNPs. This approach therefore offers a stepping stone to identification of novel genes and SNP targets linked to human longevity.
Introduction
Despite a growing number of studies on survival into old (≥ 85 years) and advanced (≥ 90 years) age, factors influencing longevity (or lifespan) are still poorly understood. Human twin studies estimated that 20–30% of variation in survival into old and advanced age, besides maintaining a healthy life style, is determined by heritable genetic factors [1, 2].
In order to determine these genetic factors, several genome-wide scans for linkage, genome-wide association studies (GWAS) and genome-wide association meta-analyses have been carried out on panels of long-lived individuals. Variations in many loci, e.g. near the D4S1564 [3], MINPP1 [4], HLA-DQA1/DRB1 and LPA [5] genes, have been identified as contributing to survival into old age, but only single nucleotide polymorphisms (SNPs) in TOMM40/APOE and FOXO3 loci were found to robustly associate with longevity [6–11]. In a whole-genome scan for genetic linkage performed by Kerber et al. [12] on individuals from the Utah Population Database, in which high levels of both familial longevity and individual longevity were exhibited, the strongest signal was observed in marker D3S3547 on chromosome 3p24.1. In addition, a locus on chromosome 3p24-22, previously identified in [13], was found to link to exceptional longevity [12], strengthening the case that genes found in these regions play a role in the regulation of human lifespan. Boyden and Kunkel [13] have identified several additional loci as having significant association with longevity, e.g. on chromosomes 9q31-34, 12q24 and 4q22-25. Recently, GWAS of parental longevity was performed on participants of European descent available via the UK Biobank [14]. Several previously known variants have been confirmed in this study. In addition, other common variants previously found by disease-specific GWAS to associate with e.g. cellular senescence, inflammation, lipid metabolism and cardiovascular conditions were also found to associate with parental longevity [14]. Their results suggest that human longevity is a highly polygenic trait influenced by many variants with a small effect size [14].
Progress in studies of human longevity is being exacerbated by small sample sizes making model organisms, such as Drosophila melanogaster, increasingly important for studying and understanding genetic factors affecting longevity. The lifespan of Drosophila is affected by several factors including genetics, differences in environmental conditions, diet and overcrowding. In laboratory conditions under controlled environment the average lifespan is found to be 26 and 33 days for female and male Drosophila, respectively [15]. Mutations in several genes have been found to increase the lifespan of Drosophila. For example, a mutation in the mth (Methuselah) G protein-coupled receptor gene, which leads to the partial loss-of-function, has been found to extend the average lifespan by 35% [16]. Mutant versions of the Indy gene, which encodes an amino acid transporter, has been shown to double the average lifespan [17]. It was also shown that single gene mutations in the target of rapamycin (TOR) and the insulin/insulin-like growth factor (IIS) signaling pathways can slow down the aging process in model organisms including flies [18].
Up to date, Drosophila GWAS have identified millions of naturally occurring SNPs that potentially influence longevity. Burke et al. [19] compared allele frequencies in the oldest surviving Drosophila with the randomly selected individuals from the same “synthetic” populations, derived from eight inbred founders. Eight significantly divergent regions have been identified. A small proportion of genes, found in these regions, were enriched in Genome Ontology (GO) biological process terms ‘defense response’ and ‘glutathione metabolic process’ [19]. Ivanov et al. [20] used lines from the Drosophila melanogaster Genetic Reference Panel (DGRP) to perform GWAS and identified ~2 M common SNPs. However, none of the SNPs found reached genome-wide significance level prompting the hypothesis of a possible combined effect of common SNPs on longevity. Gene-based analysis with either gene regions or gene regions extended into ±5 Kb of flanking sequences had identified several top-ranked genes including the CG11523 and Neprilysin 1. The former was found to have a GSK3β interaction domain that is a crucial component of the TOR pathway in human cell lines [20]; the latter could be essential for female fitness [20]. Among the top-ranked 100 genes (p < 4.79×10-6) found in this study were Chrb, slif, mipp2, dredd, RpS9 and dm genes enriched in the ’TOR pathway’ GO term [20]. Several of the longevity associated genes found are involved in processes which are known to impact aging (e.g. carbohydrate metabolism), however the function of others (although not known) provided opportunity for further, promising experimental examination. Polygenic score analysis was also used to find the additive effects of common SNPs [20]. In the absence of the second dataset, cross validation was performed. It was found that a small proportion of the observed lifespan variation (~4.7%) is explained by the additive effect of common SNPs. Despite the success in identification of variants, associated with longevity, the functional role of the majority of them – especially the variants residing outside the gene coding regions – remains to be determined.
In this paper we hypothesize that co-location of known longevity-associated genes with genes, not previously implicated in longevity, and their enrichment in the same biological function or pathway as known genes, make them novel candidate genes, potentially linked to longevity. We further hypothesize that both non-coding SNPs and their potential target genes also reside within co-located loci. To identify these novel genes/genomic regions we devised a computational approach based on analysis of networks of co-located loci, harboring both GWAS-identified variants and novel genes. Two datasets of SNPs generated by GWA studies [19–20] were used, comprising respectively ~1 million and ~2 million SNPs and sharing 2139 SNPs residing within 1515 (possibly overlapping) genes and 1044 non-coding SNPs.
As a measure of co-location (or proximity) of two distinct loci, not necessarily on the same chromosome, we used inter- and intra-chromosomal contacts generated by chromosome conformation capture Hi-C technique for the Drosophila melanogaster genome [21]. Studies of chromosome conformations have revealed that three-dimensional architecture of chromatin dictates the co-location of specific genes within the nucleus, thereby prompting the hypothesis of existence of common mechanisms controlling their transcription in a tissue-specific manner [22–23]. Recently, Won et al. [24] have demonstrated the advantages of using 3D chromatin maps for identifying target genes for schizophrenia-associated SNPs, residing within non-coding reasons of the genome. The findings have shown that for many non-coding SNPs their target genes were neither adjacent to SNPs nor in linkage disequilibrium, proving the point that many regulatory interactions are not captured by linear chromosomal organization. Analysis of intra-chromosomal interactions showed more frequent and stronger interactions within continuous genomic regions, called topologically associated domains (TADs), than with regions residing in other TADs [22–23]. TADs have been proven to play important roles in 3D organization of genomes and gene regulation and, when mutated, may lead to disease through disruption of gene regulatory pattern (reviewed in [25]).
A network of interactions was created from the inter- and intra-chromosomal contacts with nodes representing genomic regions, connected by edges, weighted by interaction frequencies. We calculated various network measures (e.g. degree [26]) and identified communities (i.e. densely connected subnetworks) existing within the network with the aim of revealing influential nodes/regions and densely connected communities (clusters) within networks. Candidate regions and communities were further explored using FlyBase (http://flybase.org/) and FlyMine (http://www.flymine.org/) resources, and GeneAge database (http://genomics.senescence.info/genes/models.html) to provide a body of evidence for genomic regions having genuine and/or previously unknown association with longevity.
To explore the role that SNPs occurring in TAD borders play in longevity, we analyzed genes residing in close proximity to TAD borders and sharing both ‘long-lived’ and ‘short-lived’ phenotypes. We hypothesized that a SNP(s) in nearby TAD borders may lead to a disruption of a regulatory pattern of a gene resulting in one of the phenotypes, ‘long-lived’ or ‘short-lived’, whereas the opposite phenotype could be a consequence of SNPs residing within genes themselves.
Results and Discussion
Choice of interaction frequency thresholds and genome-wide significance level
To assess the strength of interactions between intra- and inter-chromosomal genomic regions, distributions of interacting frequencies were analyzed individually for each chromosome and between chromosomes. Only 1% of the strongest intra-chromosomal interactions corresponding to the tails of these distributions and resulting in frequencies greater than 247, 215, 1308 and 342 for chromosomes 2, 3, 4 and X, respectively, were considered. The threshold for inter-chromosomal interaction frequencies, corresponding to 1% of strongest interactions, was 10. We refer to interactions with frequencies exceeding these thresholds as “strong” interactions.
The genome-wide significance level, required for finding association between ~106 SNPs, is usually set to p < 5×10-8. This value corresponds to 0.05 level of significance after correction for multiple testing. In our case, each SNP was binned into a 80 Kb region. There are 1503 distinct 80 Kb regions recorded in the Drosophila Hi-C data. Taking this into account, we corrected the required significance level to 3.33×10-5. In the analysis of SNPs in non-coding regions the Hi-C data with finer resolution, 10 Kb, was used where interaction frequencies between 11,839 10 Kb bins were available [21]; in this case the genome-wide level of significance was set to 0.05/11839=4.22×10-6. Following [19], SNPs with D-values exceeding 7.9 were deemed to be significant.
Original networks of interaction based on Synthetic and DGRP GWAS data
The original network of interaction based on the Synthetic GWAS data consists of 279 nodes each representing a 80 Kb region harboring at least one SNP with D > 7.9. In turn, the original network of interaction based on the DGRP GWAS data consists of 80 nodes corresponding to regions harboring SNPs with p-values < 3.33×10-5. The original networks share 14 common nodes covering 1.12 Mb of the Drosophila genome and harboring 168 genes. Only five genes ‒ Rim2 (replication in mitochondria 2), GlyP (glycogen phosphorylase), aop (anterior open), HDAC1 (histone deacetylase 1) and Tpi (triose phosphate isomerase) ‒ were found in FlyBase database as having “long-lived” phenotype. The number of SNPs residing within these common regions and satisfying chosen thresholds was 91 and 19 for Synthetic and DGRP GWAS-based data, respectively. Among the genes with the highest number of SNPs recorded in both GWAS datasets were nmo, sima, axo, CG9967, eys, chinmo and dpr3 (for the full list of genes see Supplementary Table 1).
Extended networks of interactions
Original networks were further expanded to create extended networks by adding extra nodes, corresponding to 80 Kb fragments that interact with frequencies meeting interaction frequency thresholds with the nodes, already present in the original networks. Together with regions that harbor SNPs recorded in the corresponding GWAS datasets, the extended networks contain novel regions that may not be covered by techniques used for SNP identification. We refer to these networks as Synthetic and DGRP GWAS-based (extended) networks.
The Synthetic GWAS-based extended network is fully connected and consists of 1099 nodes harboring ~75% (69,951) of SNPs recorded in the Synthetic GWAS dataset with 2,409 SNPs residing within genes. Among 13,838 genes residing within the network nodes 217 genes were found to have “long-lived” phenotype as recorded in the FlyBase database. The node labelled 547 (corresponding to region Chr2R: 20800000-20880000) has the highest degree, 150.
The DGRP GWAS-based extended network has six disconnected components and consists of 671 nodes harboring ~50% (1,093,533) of SNPs recorded in the DGRP GWAS dataset with 114 SNPs residing within genes. Among 8,929 genes residing within the network nodes 145 genes were found to have “long-lived” phenotype according to the FlyBase database. The node labelled 1183 (region Chr3R: 25920000-26000000) has the highest degree of 68.
The extended networks share 527 common nodes covering 42.16 Mb of the Drosophila genome and harboring 7,413 genes among which 121 have “long-lived” phenotype. Fifteen common regions do not harbor any genes. For approximately 30% and 3% of genes residing within common regions no SNPs were recorded in the Synthetic and DGRP GWAS datasets, respectively. Among the genes with the highest number of SNPs recorded in both GWAS datasets were Ptp61F, CG45186, kirre, Ptp99A and CG44153. Only a small proportion of genes found in regions common for both datasets were harboring SNPs meeting our significance threshold – 717 and 57 in the Synthetic and DGRP GWAS-based networks, respectively.
Several novel regions with the highest degree were selected for further analysis and each of the subnetworks centered around these novel regions (i.e. together with all connected regions) were considered (Supplementary Table 2). Genes residing within these subnetworks were sought for enrichment in longevity-associated GO terms. The results are summarized in Table 1.
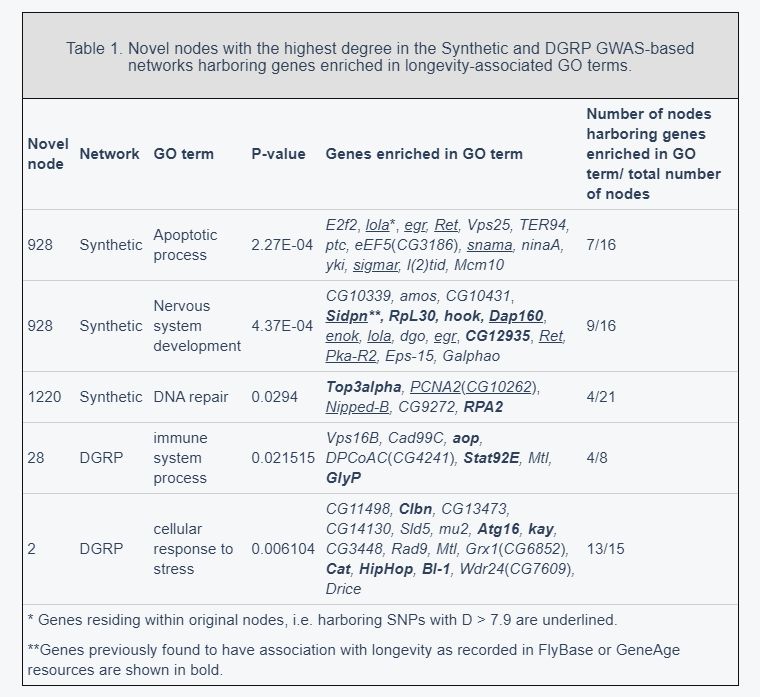
Genes residing within a subnetwork centered around node 928 (chr3R:5520000-5600000) in the extended Synthetic GWAS-based network were enriched in two GO terms, ‘apoptotic process’ and ‘nervous system development’ (Table 1). Among them the trbd and CG8412 genes that have ‘short-lived’ phenotype according to in FlyBase resources. The loss of the trbd gene, a negative regulator of the Drosophila immune-deficiency pathway, has previously been observed to reduce lifespan [27]. A number of genes in this subnetwork, including dmt, hyd, CG16908 and CG9471, were found to have phenotypes ‘increased mortality’ and ‘lethal’. The MED6 gene was found to have a phenotype of ‘cell lethal’ and is known to be required for elevated expression of a distinct set of developmentally regulated genes. This gene is essential for viability and/or proliferation of most cells and mutants of this gene have previously been observed to fail to pupate, dying in the third larval instar with severe proliferation defects in imaginal discs and other larval mitotic cells [28]. Finally, this subnetwork also contains the FoxP gene, a protein that encodes a transcription factor expressed in the nervous system. This gene has recently been shown to be important for regulating several neurodevelopmental processes and behaviors that are also related to human disease [29].
Many of the newly found genes (see Table 1) share the same biological function and co-locate with genes that have previously been reported to associate with longevity and/or aging, thus acting as a proof of concept. For example, the sidpn, hook and CG12935 genes residing in subnetwork centered around bin 928 (chr3R:5520000-5600000) were reported to have a ‘short-lived’ phenotype. Loss-of-function mutation in the hook gene has been found to reduce maximum lifespan by up to 30% [30]. Mutant flies lacking mitochondrial Top3alpha gene have also been found to have decreased maximum lifespan by up to 25%, in which a premature aging phenotype was demonstrated and mobility defects were observed [31]. Several genes, e.g. RpL30, Eps-15, Nipped-B and RPA2, listed in Table 1 were also found to have an ‘increased mortality’ phenotype according to the FlyBase resources.
Five genes residing in a subnetwork centered around bin 1220, were enriched in the ‘DNA repair’ GO term. Interestingly, this novel region is located on chr4: 960000-1040000, a chromosome seen as an anomaly because of its small size in comparison to other chromosomes and its chromatin structure. Due to its size, this chromosome is often ignored, however it is known to harbor at least 16 genes where many of them are thought to have male-related functions [32]. Using a comprehensive database of Drosophila regulatory sequences available via RedFly database (http://redfly.ccr.buffalo.edu), several enhancers were found in this region that target lncRNA sphinx and the transcription factor toy residing within this novel region although for some enhancers their target genes are not known. One can speculate that these enhancers could target genes co-located in 3D, i.e. residing within the same subnetwork centered around bin 1220.
In the extended DGRP network two novel bins, 2 and 28, were found to have the highest degree. Seven and 17 genes residing in subnetworks centered around bin 28 (chr2L: 2160000-2240000) and bin 2 (chr2L:80000-160000) were enriched in the ‘immune system process’ and ‘cellular response to stress’ GO terms, respectively. Some of these genes have previously been implicated in aging or have phenotypes which could be linked to longevity. For example, flies heterozygous for the mutation in the Stat92E gene have been found to have maximum lifespan up to 30% shorter than those of wild-type control flies [33]. The mean lifespan of Drosophila was found to be increased through post developmental RNA interference of GlyP by up to 17.1% [34]. Another gene listed in Table 1 found to have a positive effect on lifespan is Cat, where an overexpression of this gene results in an increase in lifespan by up to a third [35]. Searches in the FlyBase database show that several other genes have phenotypes associated with aging, e.g. Clbn and Atg16 genes have a ‘short-lived’ phenotype, the BI-1 gene has both a ‘short-lived’ and ‘long-lived’ phenotype and genes kay and HipHop have phenotypes for increased mortality.
Using the RedFly database, we found that the novel region on chr2L:2160000-2240000 (bin 28), which was added to the original nodes of the DGRL GWAS-based network on the basis of its strong interactions with the original nodes, harbors several enhancers. Some of these enhancers target CG34172, Uch and the transcriptional-repressor protein aop genes. The latter strongly associates with longevity and is found to be central to lifespan extension caused by reduced IIS or Ras attenuation [36]. For some enhancers their target genes were not specified. One can speculate that these enhancers could target other co-located genes residing within the subnetwork centered around bin 28.
Clusters in the extended GWAS-based networks
Community detection algorithm implemented in GEPHI which uses the Louvain modularity method [37] was performed to identify clusters in the Synthetic and DGRP GWAS-based networks. Selected clusters are shown in Figure 1. Complete sets of clusters for each network are shown in Supplementary Tables 3–4. A ‘resolution’ parameter was set to 0.1, enabling us to identify more communities/clusters as compared with the smaller number of communities that could be obtained by using a greater value for this parameter [38]. These clusters were further explored with the aim of identifying novel genes that co-locate with known longevity-associated genes and are enriched in the same biological function as known genes.
.../....
F O R T H E R E S T O F T H E S T U D Y, P L E A S E V I S I T T H E S O U R C E .
.
Edited by Engadin, 23 December 2019 - 01:19 PM.














