.
T H I S S T U D Y I S A L R E A D Y O P E N A C C E S S.
Abstract
With the ageing of the global population, interest is growing in the ‘geroscience hypothesis’, which posits that manipulation of fundamental ageing mechanisms will delay (in parallel) the appearance or severity of multiple chronic, non-communicable diseases, as these diseases share the same underlying risk factor — namely, ageing. In this context, cellular senescence has received considerable attention as a potential target in preventing or treating multiple age-related diseases and increasing healthspan. Here we review mechanisms of cellular senescence and approaches to target this pathway therapeutically using ‘senolytic’ drugs that kill senescent cells or inhibitors of the senescence-associated secretory phenotype (SASP). Furthermore, we highlight the evidence that cellular senescence has a causative role in multiple diseases associated with ageing. Finally, we focus on the role of cellular senescence in a number of endocrine diseases, including osteoporosis, metabolic syndrome and type 2 diabetes mellitus, as well as other endocrine conditions. Although much remains to be done, considerable preclinical evidence is now leading to the initiation of proof-of-concept clinical trials using senolytics for several endocrine and non-endocrine diseases.
Key points
The ‘geroscience hypothesis’ posits that manipulation of fundamental ageing mechanisms will delay the appearance or severity of multiple chronic diseases because these diseases share the same underlying risk factor — namely, ageing.
- Cellular senescence is a fundamental ageing mechanism that can contribute to or cause age-related phenotypes as well as multiple diseases, including endocrine disease, even in younger individuals (aged <40 years).
- Some senescent cells, which accumulate with ageing, develop a proinflammatory, tissue-destructive and stem cell-disrupting or progenitor cell-disrupting senescence-associated secretory phenotype (SASP), which can spread senescence to nearby and distant non-senescent cells.
- Senolytic agents have been discovered that selectively eliminate senescent cells by transiently disabling the survival pathways (senescent cell antiapoptotic pathways) that protect senescent cells against their own SASP.
- Preclinical studies suggest that senolytics hold promise for delaying, preventing or treating many age-associated disorders.
- Despite the growing experimental support for targeting cellular senescence to treat multiple age-associated diseases simultaneously, carefully conducted clinical trials in humans are needed to better define the benefits and risks of senolytic drugs.
Introduction
Ageing is now generally accepted as the single largest risk factor for many of the major chronic diseases (for example, type 2 diabetes mellitus (T2DM), cardiovascular disease and cancer) that account for the bulk of morbidity, deaths and health costs in the USA and other developed countries1. For these conditions, the predictive ability of advanced chronological age exceeds the predictive ability of all other risk factors combined. Enormous progress has been made over the years in the development of specific drugs to treat individual diseases associated with ageing, including metabolic dysfunction and T2DM (for example, GLP1R agonists and DPP4 inhibitors), skeletal fragility and osteoporosis (for example, bisphosphonates and denosumab) and vascular dysfunction and disease (for example, statins), with potential new drugs on the horizon to treat skeletal muscle loss and frailty (for example, the activin type 2 receptor antibody bimagrumab); however, the combined effect of these drugs on reducing morbidity and mortality has been modest.
Age-associated chronic diseases rarely, if ever, exist in isolation in elderly individuals. Rather, they tend to occur in synchrony as multimorbidities, the prevalence of which increases exponentially after age 70 years2. This multimorbidity leads to substantial barriers to the appropriate treatment of each disease, including prioritization of each condition by the busy primary care physician and perhaps most importantly the growing problem of polypharmacy in older (aged >60 years) patients. Indeed, the most recent estimates indicate that 36–39% of adults aged 62 years or older take five or more prescription medicines3. When over-the-counter medication use is included, the prevalence of such adults taking five or more medications increases to 67% (ref.3). Fundamentally, polypharmacy occurs because most treatment strategies for chronic age-related morbidities are disease specific. This specificity inevitably leads to polypharmacy, which can result in problems related to adverse effects, unpredictable drug interactions and poor adherence.
Osteoporosis provides a good case study of the reasons why a disease-specific approach fails in older (aged >60 years) patients with multimorbidities. Currently, numerous options are available for the treatment of osteoporosis, including a selective oestrogen receptor modulator (raloxifene), four bisphosphonates (alendronate, risedronate, ibandronate and zoledronic acid), a human monoclonal antibody to receptor activator of nuclear factor-κB ligand (RANKL; denosumab), analogues for parathyroid hormone and parathyroid hormone-related protein (teriparatide and abaloparatide) and a monoclonal antibody to sclerostin (romosozumab)4. Despite these treatment options, most patients with osteoporosis, including following an event as devastating as hip fracture, remain untreated5. A number of reasons could explain the lack of appropriate treatment for age-associated co-morbidities, including osteoporosis, some of which are condition specific. For example, in osteoporosis, the fear of rare bisphosphonate-related adverse effects, such as osteonecrosis of the jaw or atypical femur fractures6, can reduce treatment uptake.
Within the context of multiple co-morbidities of ageing and the growing recognition by the gerontology community that ageing in itself is the largest risk factor for most age-related chronic diseases, the ‘geroscience hypothesis’ has gained accelerating momentum. This hypothesis posits that manipulation of fundamental mechanisms of ageing will delay (in parallel) the appearance or severity of multiple chronic diseases because these diseases share the same underlying risk factor — namely, ageing7 (Fig. 1). The potential efficiency of targeting fundamental ageing mechanisms, as opposed to treating each age-associated condition separately, is truly remarkable. Indeed, by one estimate, a 2% delay in the progression of ageing processes would lead to an increase of ten million healthy (as opposed to disabled) elderly people in the USA by 2060 compared with doing nothing, which would delay the onset of cancer or heart disease8. This increase would correspond to savings in US health costs of $7.1 trillion over 50 years8.
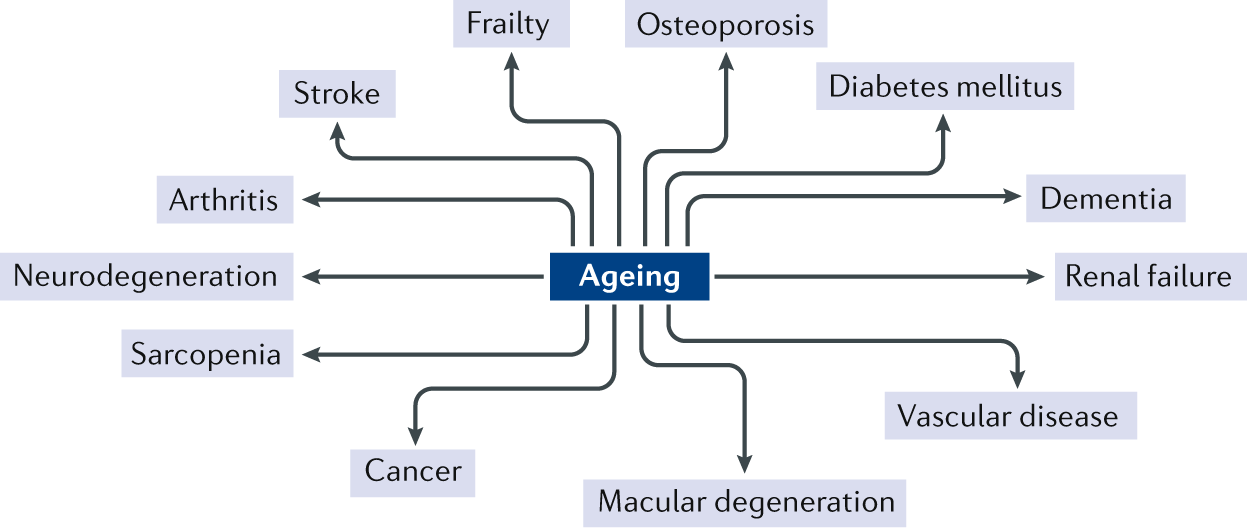
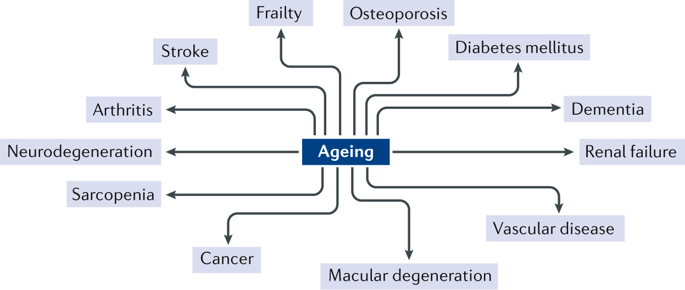
This figure shows examples of chronic non-communicable diseases that have ageing as one of the main risk factors. Reprinted with permission from ref.11, JCI.
A number of common ageing mechanisms that influence lifespan and healthspan have been identified on the basis of studies across a range of species9. These ageing mechanisms can be categorized into nine hallmarks, which are genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication9 (Fig. 2). Although it is useful to consider these hallmarks individually in mechanistic studies related to ageing, they are highly interconnected, and, ultimately, systems biology approaches will be needed to fully understand interactions among these pathways as they contribute to ageing. Of these fundamental ageing mechanisms, cellular senescence has received considerable attention as a process that is potentially druggable to prevent or treat multiple ageing comorbidities.
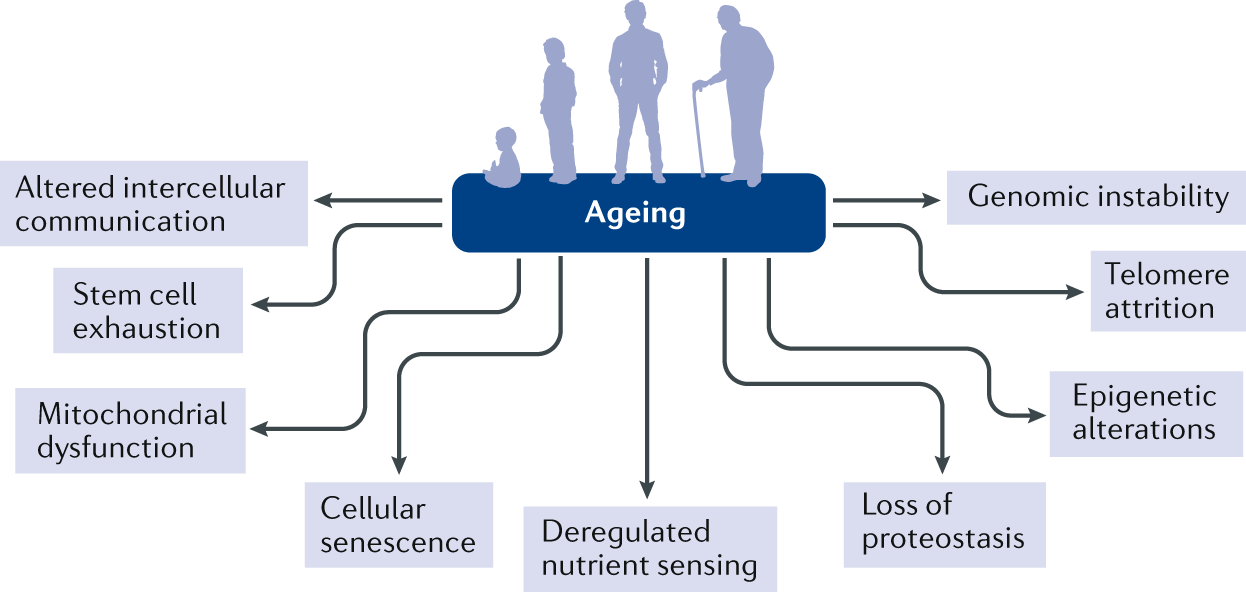
Nine fundamental physiological, cellular and molecular hallmarks of ageing that provide a useful framework for mechanistic studies. Reprinted with permission from ref.9, Elsevier.
In this Review, we discuss the biological mechanisms of cellular senescence and the evidence that this physiological process has a causative role in multiple diseases associated with ageing. Subsequently, we focus on the role of cellular senescence in a number of endocrine diseases, as well as approaches to target this mechanism to prevent or alleviate age-associated endocrine diseases.
Mechanisms of senescence
Cellular senescence, originally described by Hayflick10, is a cell fate that involves essentially irreversible replicative arrest, tumour suppressor activation, profound chromatin changes, apoptosis resistance and frequently increased protein synthesis11. The increase in protein synthesis includes excessive production of proinflammatory cytokines (the senescence-associated secretory phenotype (SASP)), which is responsible for tissue damage and contributes to ageing across tissues11.
Signals for senescence
Multiple signals can induce a cell to enter senescence (Fig. 3). These include external signals, such as damage and/or danger signals (for example, damage-associated molecular patterns or pathogen-associated molecular patterns), metabolic signals (for example, high levels of glucose, ceramides, certain fatty acids, prostanoids, hypoxia and reactive oxygen species (ROS)), factors that induce repeated cell replication (for example, growth hormone or IGF1), inflammatory factors, radiation, shear or mechanical stress, certain extracellular vesicles and other factors (for example, signals originating from other senescent cells that lead to spread of senescence)11,12,13,14,15,16,17,18,19,20,21,22,23. Internal factors that can drive cells into senescence include telomeric dysfunction from repeated cell replication, DNA damage, aneuploidy, oncogene activation, mitochondrial dysfunction, intracellular metabolites (for example, ROS), nuclear envelope dysfunction and certain genetic variants or mutations (for example, those involving lamin A, DBC1 or p53)11,15,16,22,24,25,26,27,28,29,30,31. These senescence-inducing signals activate transcription factor cascades, including pathways involving the cell cycle inhibitors p16INK4A/RB and p53/p21CIP1.
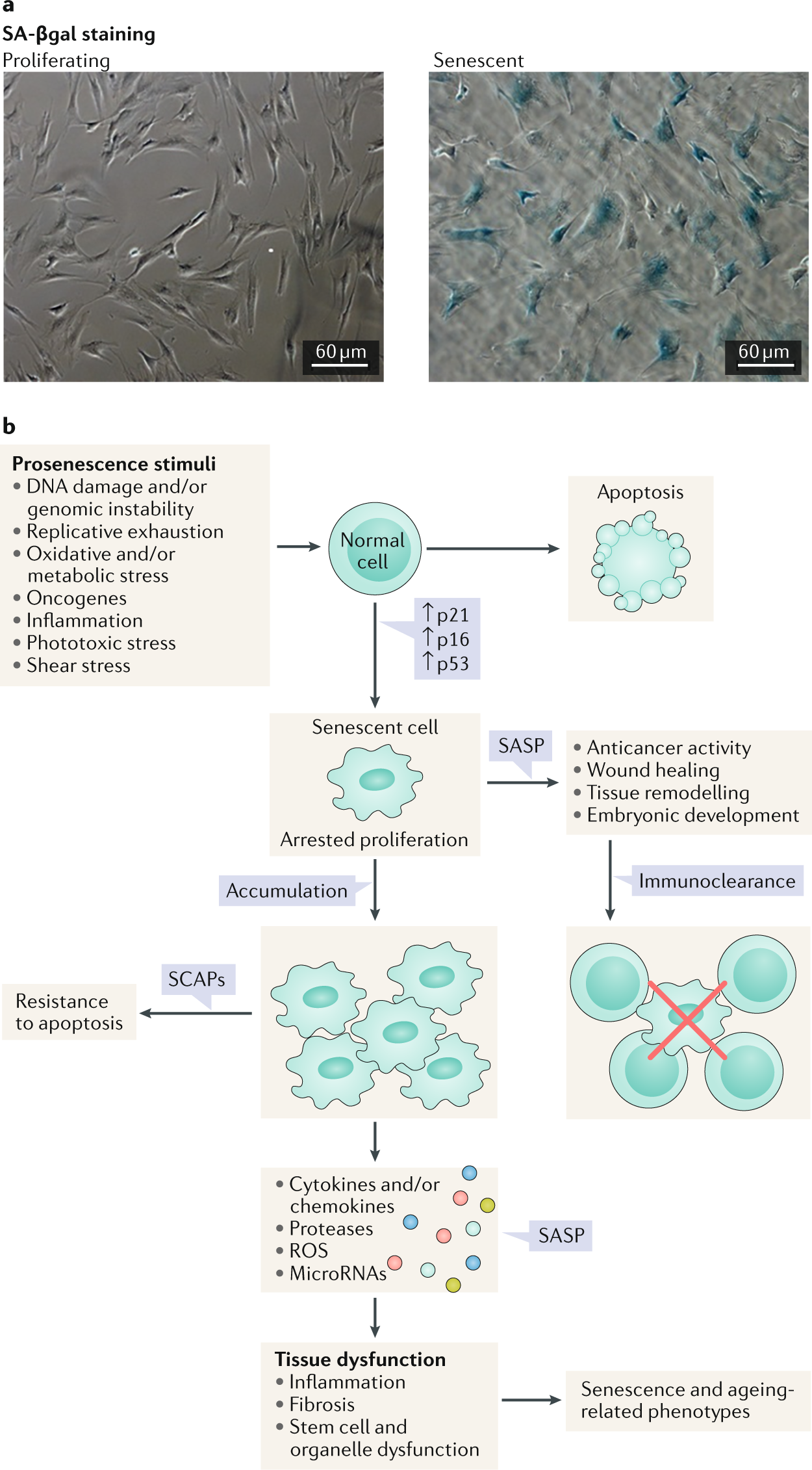
a | Images of senescence-associated β-galactosidase (SA-βgal) staining of proliferating and senescent human preadipocytic cells. Bars represent 60 µm. b | Different stimuli can induce cellular senescence. The phenotypes of senescent cells are cell type and context dependent; however, cells can develop a senescence-associated secretory phenotype (SASP), arrested proliferation and resistance to proapoptotic pathways through senescence-associated antiapoptotic pathways (SCAPs). The formation of senescent cells in response to damage has an important role in tumour suppression and is also necessary for tissue repair (for instance during wound healing). By contrast, the persistent presence of fairly small numbers of senescent cells can promote a protumorigenic milieu and impair the function of multiple tissues. ROS, reactive oxygen species.
The senescent phenotype
At least in cell culture, 2 weeks or more after the initiation of senescence, the senescent cell phenotype can become established and essentially irreversible. Cellular senescence is characterized by loss of proliferative potential, changes in expression of multiple genes, altered histone binding, changes in the levels of heterochromatin and/or euchromatin, resistance to apoptosis, increased metabolic activity, increased protein production and frequently a SASP11. The SASP involves secretion of inflammatory cytokines, chemokines that attract and anchor immune cells, tissue-destroying proteases, factors that affect stem and progenitor cell function (such as activin A), haemostatic factors (such as plasminogen-activated inhibitor 1), vasopressors, ceramides, prostanoids, bradykinins, nucleotides (such as microRNAs, mitochondrial DNA and nuclear DNA) and growth factors. These factors can act in a paracrine or endocrine manner to induce local and systemic inflammation, immune system activation, fibrosis, apoptosis, tissue damage, and stem and progenitor cell dysfunction and can induce the spread of senescence to other cells both locally and systemically11,15,16,26,32,33,34 (Fig. 3).
Senescent cells can undergo a partial Warburg shift with increased glycolytic metabolism, reduced fatty acid utilization, intracellular accumulation of lipids and increased ROS generation, which perturbs nearby cells and contributes to the spread of senescence to non-senescent cells in vitro and in vivo34,35. Several months after cells have entered senescence, transposons (such as LINE1 and other elements) can be produced within senescent cells along with reverse transcriptase. These elements lead to DNA rearrangements within the cell and further activation of DNA damage signals that can induce intracellular cGAS–STING damage-response pathways, thereby reinforcing and further exacerbating the proinflammatory, proapoptotic SASP36.
The composition of the SASP differs depending on the cell type from which the senescent cells originated, the specific inducers that caused senescence and the hormonal milieu. For example, transcriptome analysis of senescent fibroblasts from different tissues showed considerable heterogeneity in the SASP37. Furthermore, the SASP varies over time and seems to be modifiable: hormones and drugs such as glucocorticoids, rapamycin, metformin or JAK1/JAK2 inhibitors can inhibit components of the SASP both in vitro and in vivo26,38,39,40,41. In addition, external signals can upregulate the SASP. For example, at least in vitro, tumour necrosis factor (TNF) can exacerbate and reinforce the pro-inflammatory nature of the SASP14.
Markers of cellular senescence
Much more research needs to be conducted on developing and optimizing sensitive and specific assays that track changes in the burden of senescent cells using biopsy samples, blood assays, assays of other body fluids or imaging42. Complicating such work, the definition of cellular senescence is somewhat vague: several potentially proinflammatory cell types, such as macrophages, osteoclasts or precancerous or cancer cells, share many features with senescent cells43. Furthermore, these proinflammatory ‘senescent-like’ cells could arguably even be the same as what are currently regarded as being senescent cells43. Few cellular or tissue markers are highly sensitive or specific for senescent cells, so combinations of these markers generally need to be analysed to draw firm conclusions about the effects of diseases or interventions on the burden of senescent cells. These in vitro and in vivo markers of senescent cells include increased cell size and intracellular protein content, accumulation of lipofuscin, increased expression of p16INK4A, p21CIP1 and SASP factors (for example IL-6, IL-1α, IL-8, monocyte chemoattractant protein 1, plasminogen-activated inhibitor 1 and plasminogen-activated inhibitor 2), increased cellular senescence-associated β-galactosidase (SA-βgal) activity, cytoplasmic HMGB1, the presence of senescence-associated distension of satellites (SADS) and telomere-associated DNA damage foci, among other markers44,45,46,47,48.
These markers of cellular senescence have their limitations. For example, both SA-βgal activity and p16INK4a expression can be high in activated macrophages, and p16INK4A expression can also be elevated in certain differentiated cell types in vitro and in vivo43,49. Furthermore, levels of p16INK4A are not elevated in all cells that could be considered to be senescent50. Thus, SA-βgal activity and p16INK4a expression are neither fully sensitive nor specific markers of senescent cells. A combination of assays is needed to determine senescent cell numbers in tissue samples.
Currently, it is unclear if circulating SASP factors or local senescent cell numbers in biopsy samples of skin, adipose tissue or other tissues reflect the overall burden of senescent cells. New or composite assays are needed for use in clinical trials, for example assays of circulating or urinary microvesicles originating from senescent cells, senescence-specific microRNAs or other nucleotides, or epigenetic profiles. Imaging methods also need to be developed to follow localized populations of senescent cells, for example in trials of senolytic drugs for idiopathic pulmonary fibrosis51 or osteoarthritis52.
Physiological relevance of cellular senescence
Consistent with the notion of cellular senescence being a terminal cell fate that occurs in response to certain stimuli, senescent cells can develop at any point during life. For example, senescent cells can even occur in 32-cell-stage human blastocysts in response to hypoxia53. However, studies conducted using combinations of senescence markers suggest that the abundance of senescent cells increases with ageing in multiple tissues of mice, monkeys and humans20,54,55,56. In otherwise healthy humans, the abundance of senescent cells can increase from low or even nearly undetectable levels in skin or adipose tissue biopsy samples from young individuals (that is, aged 20–30 years) to as many as 5% of cells or even more in individuals aged 40–80 years; however, more research using a broader range of markers of cellular senescence needs to be undertaken57,58,59,60. Consistent with a relationship between cellular senescence and ageing, interventions that increase healthspan and lifespan, including caloric restriction or mutations in the growth hormone axis (for example, those in Ames dwarf mice (which lack growth hormone) and growth hormone receptor gene-knockout mice) are associated with decreased burden of senescent cells in mice54,61.
Senescent cells that have a proinflammatory SASP can cause substantial pathogenic effects, resulting in phenotypes that are associated with ageing. For example, transplanting small numbers of senescent cells (mouse fibroblasts induced to become senescent by radiation) around the knee joints of young mice induces an age-related osteoarthritis-like condition52. Furthermore, transplanting autologous ear fibroblasts or syngeneic (genetically compatible) preadipocytes induced to become senescent by radiation or chemotherapeutic drug exposure intraperitoneally into lean middle-aged mice is sufficient to cause frailty, early onset of all age-related diseases and premature death compared with transplanting similar numbers of non-senescent cells. This effect occurs even though only 1 in 10,000 of the total number of cells in the recipient mice is a transplanted senescent cell34. Moreover, transplanting only half the number of senescent cells was sufficient to cause an early ageing phenotype and increased mortality in middle-aged diet-induced obese mice or lean old mice as opposed to lean middle-aged mice34. Thus, even small numbers of senescent cells can cause considerable dysfunction.
Targeting cellular senescence
Senescent cells can be beneficial in certain circumstances: for example in the defence against cancers, in the course of wound healing or in placental regulation of parturition62,63,64. Hence, interfering with the generation of new senescent cells is probably not a viable overall therapeutic strategy. For example, systemically reducing the expression of p16INK4A or p53 might delay the development of senescent cells but might also increase the risk of cancer63. However, once formed, senescent cells harbour oncogenic mutations and cause dysfunction owing to their SASP. Removing these already formed and persistent senescent cells might decrease cancer risk, decrease the spread of senescence and reduce tissue dysfunction.