































.
F U L L T E X T S O U R C E : Genome Biology
Abstract
Epigenetic clocks comprise a set of CpG sites whose DNA methylation levels measure subject age. These clocks are acknowledged as a highly accurate molecular correlate of chronological age in humans and other vertebrates. Also, extensive research is aimed at their potential to quantify biological aging rates and test longevity or rejuvenating interventions. Here, we discuss key challenges to understand clock mechanisms and biomarker utility. This requires dissecting the drivers and regulators of age-related changes in single-cell, tissue- and disease-specific models, as well as exploring other epigenomic marks, longitudinal and diverse population studies, and non-human models. We also highlight important ethical issues in forensic age determination and predicting the trajectory of biological aging in an individual.
Introduction
A key question in biology is to understand why and how we age. Alongside this, the unprecedented gain in the average lifespan in humans, since the mid-twentieth century, has dramatically increased both the number of older people and their proportion in the population. This demographic phenomenon is changing our societal make-up, from only ~130 million being 65 years or older (~5% of the world population) in 1950, to a predicted ~1.6 billion people (~17%) by 2050 [1]. However, the success in reducing mortality has not been matched with a reduction in chronic disease [2]. This leads to the undesirable outcome of many years of this prolonged lifespan being spent in ill health, with an associated massive health care burden. Increasing the productivity and reducing the disease affliction in these extended years7 would be clearly beneficial for both the individual and society [2, 3]. This aim of maximizing the “healthspan” [2] makes obtaining accurate measures of aging-related pathology essential, to gauge its speed, decipher the changes that occur, and potentially unlock how aging acts as a disease risk factor [4]. There is considerable population variation in the rate at which people visibly age [5] as well as become impaired by age-related frailty and disease [2]. Measurement of this relative “biological” aging [2] may allow pre-emptive targeted health-promoting interventions, perhaps in a personalized and disease-specific fashion. It would also aid in testing interventions that attempt to modulate the aging process [6].
The cellular and molecular hallmarks of aging include changes associated with cell senescence, dysregulated nutrient sensing, and stem cell exhaustion, among others [6]. Therefore, many biological measures, such as p16ink4a tissue levels, circulating CRP, creatinine, and fasting glucose, as well as telomere length all correlate with aging [6,7,8]. In this decade, we have discovered the remarkable power of epigenetic changes to estimate an individual’s age [9, 10]. Epigenetics encapsulates the chemical modifications and packaging of the genome that influence or indicate its activity [11], with strict definitions requiring inheritance through mitotic cell division [12]. Observations of age impacting on this mechanism have been reported for more than 50 years [13,14,15,16] and suggested a role in age-related disease [17]. However, the association between epigenetic modifications and age became most starkly apparent with the arrival of the first high-throughput arrays measuring DNA methylation [18,19,20]. These high-resolution data enabled the construction of extremely accurate age estimators, termed “Epigenetic” or “DNA methylation clocks” [21,22,23,24,25]. Subsequently, these clocks were reported to capture aspects of biological aging and its associated morbidity and mortality [26,27,28,29]. DNA methylation (5′methylcytosine, 5mC) is the most common DNA modification and predominantly occurs at cytosines in a CpG dinucleotide context in differentiated mammalian cells. The stability of 5mC in biological samples, even from long-term stored DNA, brings large-scale data availability, for use in subsequent high-throughput analysis.
In this paper, we discuss the scientific challenges that the fascinating discovery of “DNA methylation clocks” has brought into focus. We provide recommendations and suggest future experiments required to dissect the strengths and weaknesses of this important biomarker, in order to probe its biological significance, cellular mechanics, and epidemiological potential. We do not review in depth the background history and current state of the clocks themselves; we refer readers to recent excellent reviews for this information [9, 10]. Instead, the purpose is forward-focused, i.e., to define the current issues, to suggest what will aid unlocking future potential, and to further explore and define any functionality, with the hopeful long-term benefit of increasing the “healthspan.”
Here, we define a “DNA methylation clock” as an estimator built from epigenetic DNA methylation marks that are strongly correlated (r ≥ 0.8 [9]) with chronological age or time, which can accurately quantify an age-related phenotype or outcome, or both. These DNA methylation clocks are generally built with a supervised machine learning method, such as a penalized regression (e.g., lasso or elastic net) trained against chronological age to identify an informative and sparse predictive set of CpGs [9, 10]. The residual, or error from chronological age, is used as a marker for biological age of an individual [9, 10]. The age-related phenotype or outcome may be disease, mortality, clinical measures of “frailty,” or cellular phenotypes, including the mitotic age (the total number of lifetime cell divisions of a tissue [30, 31]).
It is evident, even from our initial observations so far, that the aging-related epigenetic modifications captured by DNA methylation clocks are pervasive and indicative of genomic, cell biology, and tissue changes occurring over the life-course. These molecular alterations may bring a high-resolution and precise understanding of age-related pathology and physiology.
Challenge 1
Delineation of the chronological and biological components of DNA methylation clocks
Current knowledge
DNA methylation-derived epigenetic clocks are currently better in estimating actual chronological age than transcriptomic and proteomic data, or telomere length [7]. However, it was recognized that some variability in these initial clocks’ age estimation existed, which was identified to be a measure capturing individual variation in biological age. Age acceleration, defined as the difference between this epigenetically measured age and the actual chronological age, was associated with mortality [26] and other age-related phenotypes or diseases [32,33,34,35,36,37,38,39].
Of the first-reported clocks, the Hannum et al. clock was trained and tested on blood-derived DNA [23]. It comprises 71 CpG selected from the Illumina 450k array that strongly capture changes in chronological age, which is partly driven by age-related shifts in blood cell composition [23]. The Horvath clock was constructed across multiple tissues, including the blood data from Hannum et al., as a potential “pan-tissue” master clock of chronological age, and focused on capturing shared changes, independent of tissue type [24]. It included 353 CpGs that were present on the earlier generation Illumina 27k array. These differences in training sets led to some conflicting findings between reported associations [7, 29, 40].
Aging leads to epigenetic alterations, including changes in DNA methylation, through both multiple distinct and intersecting age-related mechanisms [6, 41]. Many DNA methylation aging clocks have now been derived, and due to their individual strengths and weaknesses, explicit reference must be made to the specific clock employed (see further in “Challenge 2”). Captured age-related epigenetic variation can be firstly split into intrinsic, or intra-cellular, and extrinsic, or broadly within-tissue and external, aspects of the aging process [27]. The former is a surrogate readout of multiple cellular and genomic processes, including possible deterioration of mechanisms involved in maintaining the epigenome, while the latter includes age-related cell proportion changes within a tissue. While these first clocks are markers capturing these effects to a greater or lesser extent [42], both can predict all-cause mortality at a population, but not individual level, even after correcting for known risk factors [27]. To investigate biological age more directly, clocks have also been trained on age-related and disease phenotypes in combination with chronological age, such as the “PhenoAge” DNA methylation clock that incorporates nine age-related biochemical measures [43]. Cigarette smoking, a significant disease-related factor, is observed to strongly drive mortality-associated predictive DNA methylation changes [44]. However, these tobacco-related methylation changes do not influence the Horvath or Hannum et al. clocks, but are captured in “PhenoAge” [9]. Of note, a very recently constructed mortality predictive DNA methylation clock, termed “GrimAge,” directly incorporates smoking-related changes through an estimate of “pack-years” smoking. This clock also includes certain plasma protein levels estimated by DNA methylation, and this leads to an even stronger prediction of both lifespan and healthspan [45].
Current uncertainty
The first DNA methylation clocks devised were found to be useful for estimating actual age, as well as capturing associations with biological aspects of aging. Data gathered from these early clocks can still be exploited for both these chronological and biological measures. However, now this duality has been recognized, we can attempt to improve our assessment of these two characteristics. Specialized clocks are likely to be more powerful for accurate age prediction or to capture specific biological aging-related functional deterioration or disease-related predictions [45]. How far these two distinct uses can be separated into discrete clocks and improved for their specific role is presently unknown. However, clearly if the DNA methylation clock measurement of actual age was perfect, the loss of any variability removes the window where biological aging associations can be made [46]. Empirical calculations estimate that near-perfect forensic age determination may be possible with large enough sample size, even with current DNA methylation array platforms (see Fig. 1a) [46], although this statistically derived view that chronological clocks can approach extreme precision is not held by all in the field.
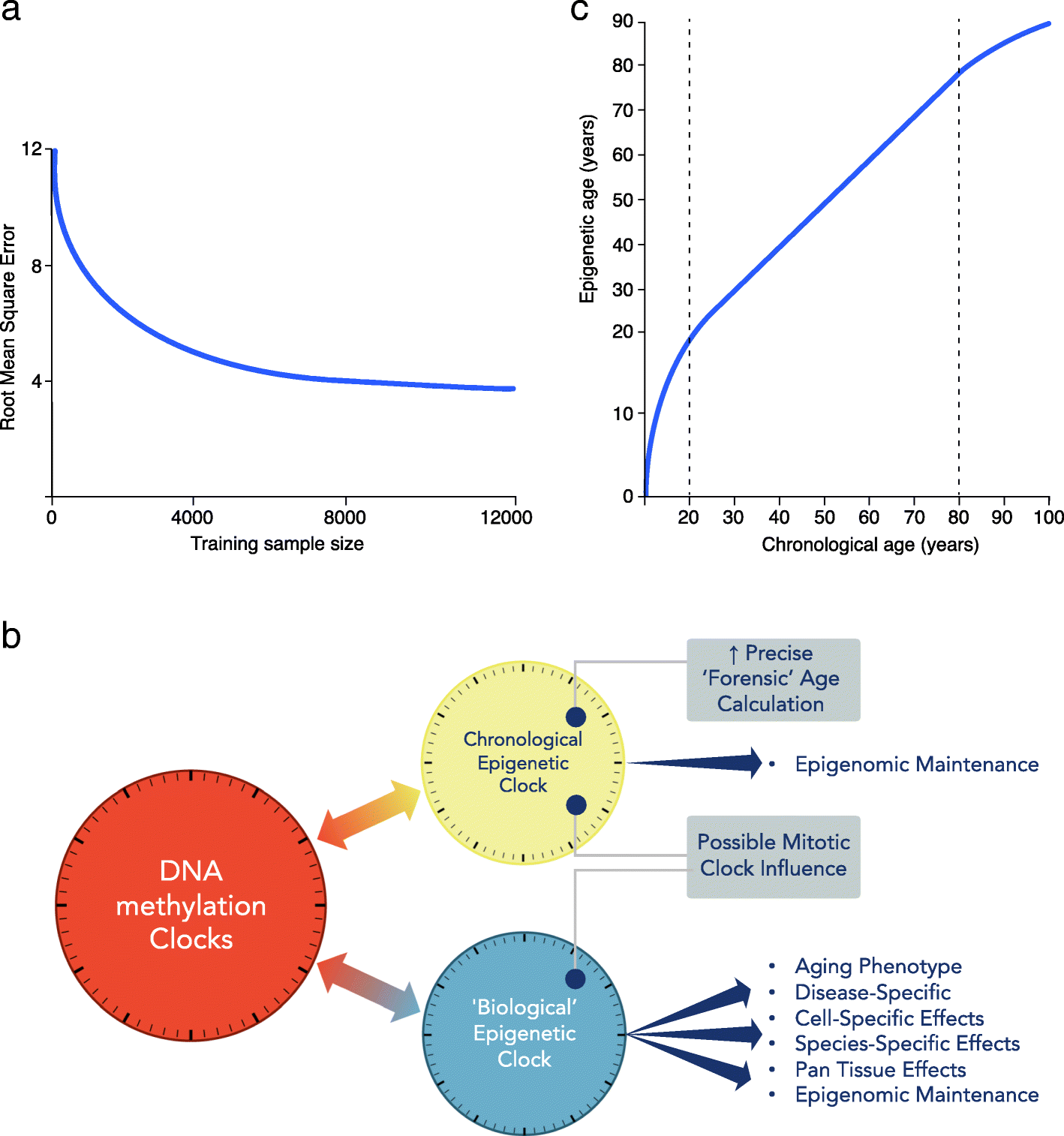
a Chronological age estimation error. With increasing training sample size, improved measurement of chronological age is expected, even using current array data (adapted from Zhang et al. [46]). y-axis: root mean square error (RMSE) of the predicted age. b DNA methylation clocks contain both chronological and biological information. The relative proportions of each will depend on the CpG probes employed in the construction of the clock. Therefore, there are multiple clocks that can be deconvoluted from aging-related epigenetic changes. Moving forward, more precise chronological (forensic age clock) and biological clocks, specific for particular diseases, informative of health or disease state need to be defined and separated. c Epigenetic age trajectory. Epigenetic age is not linear over the life course. Chronological age in years (x-axis) and epigenetic age in years (y-axis)
Each DNA methylation clock that is constructed is unique to its method of calibration [47], indicating the importance of tissue/s employed, number of samples, and statistical methodology. Clearly, small sample sizes are more susceptible to multiple aging-related confounders, measurement errors, and imperfect statistical predictions. Even when clocks are directly trained on actual chronological age, the strong influence of age-related biological processes may skew the CpGs selected for the clock, underscoring the importance of an appropriate population of sample donors. Furthermore, as discussed in “Challenge 3,” Zhang et al. recently highlighted the impact of not only sample size but also cell type correction, in heterogeneous cell type-derived DNA, on improving chronological age prediction [46].
For “Biological” clocks, another obvious area of uncertainty is that there is not one measure or “gold standard” of biological aging [6, 7, 41, 48]. This phenomenon encompasses a wide range of age-associated changes from the merely visible to disease-risk related. To understand how aging may be characterized by chronological and biological age-related epigenetic changes, we need more detailed understanding of what mechanisms may be underlying these observations. There is no evidence that the Horvath or Hannum et al. clock CpGs are enriched for functionality over and above the promoter-focused arrays from which they were constructed. Furthermore, the clocks have shown variability in their ability to capture measures of mitotic age, such as telomere length [9, 49], due to their differing training models. In general, epigenetic aging is distinct from senescence-mediated aging and is not prevented by telomerase expression [50,51,52]. A recent DNA methylation telomere clock identified that although this clock was trained on telomere length, it more strongly reflected cell replication and, moreover, associated with aging-related phenotypes more strongly than telomere length itself [53].
Future experiments and recommendations
Understanding the chronological and biological drivers of these DNA methylation clocks will require them to be teased apart as much as possible (see Fig. 1b). The clear separation between these two factors, down to specific sets of CpGs, would lead to more powerful specialized clocks and distinct mechanistic studies.
To obtain the most precise estimate of actual age and quantification of its robustness from easily assessable DNA requires appropriately powered large-scale DNA methylation analyses. This is especially the case if this measure is to be used as a legal measure of human age [54,55,56,57]. Testing across the range of routinely collected DNA samples will be needed, such as those gathered from peripheral blood or buccal swabs, but also other sources of DNA, such as hair root, skin, and other tissues. However, this is currently only likely to be tractable in data derived from peripheral blood, as these are available at large scale. For the other tissues, the approach is likely to be insufficiently powered in the intermediate future. Specific CpGs will be selected to construct clocks for high-precision forensic age estimation, when chronological age is not known or disputed. They will employ those CpGs that are the most robust and accurate for particular tissues and their constituent cell types [58]. We will need to define the influence of genetic variation and environmental factors on these measures. Accumulating this knowledge of the various DNA methylation clocks will guide their future legal or forensic application [59].
The biological aging component captured by epigenetic age acceleration consists of a large range of drivers, including tissue-specific, cellular aging pathology, stochastic deterioration, and disease-related factors. As mentioned, there is no single measure of biological age; therefore, specific components of aging biology should be focused on and interrogated. This includes aging-related biological pathways involving, for example, mTOR, IGF-1, and p53 [6], as well as epigenomic aspects including the polycomb repressive complex, TET/DNMT levels, and H3K36 methylation [60, 61] (see Table 1). This refined analysis could bring new molecular mechanistic insight to the aging process.
.../...
.
Edited by Engadin, 30 November 2019 - 05:00 PM.














