































.
Posted today by user "Nigeria Custom Officer"
O P E N A C C E S S S O U R C E : AIMS Molecular Science
1. Introduction
From current studies on human whole genomes, we have already learned that “cancer is a genetic disease” after understanding the genomic/genetic differences between cancer and normal cells through next-generation sequencing [1,2]. At present, whole-genome sequencing data can predict the incidence of specific diseases, including cancer, in healthy individuals [3]. However, recent metabolome studies suggest that metabolites could be biomarkers for cancer and its development [4], suggesting as well that “cancer is a metabolic disease”. It is well known that cancer mainly uses glucose to produce adenosine triphosphate (ATP) by glycolysis. The “Warburg effect”, or abnormal metabolism, which was described by Dr. Otto Warburg over 60 years ago, is widely known as one of the most essential characteristics in cancer cells [5,6,7]. Importantly, it is frequently observed that both dysregulation of the tricarboxylic acid (TCA) cycle (Krebs/Citrate cycle) and oxidative phosphorylation occur in cancer cells, suggesting that mitochondrial dysfunction precedes the metabolic change [8], indicating that “cancer is a mitochondrial disease”. Mitochondria have their own DNA or mitochondrial DNA (mtDNA), suggesting that they have evolved from the symbiosis of aerobic bacteria (α-proteobacteria) into ancestral eukaryotic cells. However, it should be noted that most (99%) of the genes that encode mitochondrial function-associated proteins are contained in nuclear genomes [9]. Therefore, mitochondrial function-associated genes are commonly regulated by transcription factors (TFs) that regulate other cellular protein-encoding genes.
In this regard, scientific interest should be focused on causative factors that lead to mitochondrial dysfunction by asking whether “cancer could be a transcriptional disease”. In this chapter, through a discussion mainly about biological functions and regulations of nicotinamide adenine dinucleotide (NAD+), which is not only required as a co-enzyme for various biologically significant reactions but also as a substrate for poly (ADP-ribose) polymerase (PARP), we propose a novel anti-cancer therapy that converts or forces cancerous cells to return to a non-proliferating or differentiated state.
2. Differences in metabolism between cancer and normal cells
Before discussing the biological roles and functions of the NAD+ molecule, we should reconfirm the characteristics of cancer cells, which frequently show differences in metabolism and transcriptional state from normal healthy cells. Cancer cells very frequently show disruption in metabolism compared with normal differentiated cells. First, proliferating mammalian cells exhibit anaerobic metabolism [6], and the energy they require is thought to be supplied mainly from glycolysis. Moreover, in cancer cells, abnormalities in structures and functions of mitochondria are observed [8]. Hence, deficiencies in carbohydrate metabolism, including the TCA cycle and oxidative phosphorylation, which are carried out in the mitochondria, are frequently accompanied by malignancies of cancer cells [7]. Second is the difference in amino acid metabolism. It is well known that cancer cells require high amounts of glutamine to survive and proliferate [10,11,12]. Expression of the GS and GDH genes, which encode glutamine synthetase and glutamate dehydrogenase, respectively, is significantly increased in breast cancer cells, and accumulated ammonia is efficiently used for glutamine and glutamate synthesis without engaging the urea (ornithine) cycle [13]. As described in many textbooks on biological chemistry, the TCA cycle is overlapped by the urea cycle, in which ammonia is processed to urea and is safely eliminated. However, in cancer cells, the urea cycle does not work efficiently because of insufficient progression of the TCA cycle. In addition, serine and one-carbon metabolism is also dysregulated in cancer, which might also affect the rate of synthesis of thymidine and purine nucleotides [14]. If mitochondrial functions are disrupted, serine synthesis, which is catalyzed by SHMT2 with the coenzyme tetrahydrofolate, would be insufficient in the mitochondria [14]. Consequently, cancer cells might exhibit dependence on higher amounts of serine to produce more glycine for use in the de novo synthesis of purines and pyrimidines. Generally, de novo synthesis of nucleotides is hyperactivated in proliferating cells. Moreover, malignant tumors usually utilize greater amounts of uracil compared with the normal surrounding cells [15]. Thus, the nucleotide metabolism in cancer cells is significantly different from that in normal cells [16,17]. In proliferating malignant cancer cells, both higher telomerase reverse transcriptase activities and longer telomeric regions of chromosomes than in normal differentiated cells are observed [18]. Nucleotides that were synthesized in excess could be incorporated into the terminal regions of chromosomes that will result in telomere elongation. This may be consistent with the observations that communications between mitochondria and nuclear DNAs/telomeres play roles in the regulation of longevity and the incidence of cancer [19,20].
It is widely known that cancer cells carry numerous genomic mutations in chromosomal DNAs. Recent studies on human genomic DNA sequences revealed several cancer predisposition genes [2]. In this context, “cancer is a genetic disease” [21]. However, cancer cells have many different characteristics of metabolism from those of normal differentiated cells, including those related to carbohydrates, amino acids, and nucleotides. Because most of these biological reactions depend on mitochondrial functions, “cancer is a metabolic/mitochondrial disease” [8]. Although mutations in mtDNA cause mitochondrial deficiencies that could lead to cancer development [22], most of the mitochondrial proteins, including the TCA cycle/urea cycle enzymes, are encoded on nuclear DNAs [9]. Not only cancer but also neurodegeneration is thought to be caused by mitochondrial deficiencies [23]. To better understand molecular pathogenesis in the development of cancer and aging-related diseases, transcription mechanisms that control the expression of mitochondrial function-associated genes should first be elucidated.
3. Niacin (vitamin B3) and biological functions of NAD+
Niacin is classified as vitamin B3, whose depletion causes pellagra. As shown in Figure 1, nicotinic acid (NA) and nicotinamide, which provide the biological activity of niacin, are generated from the metabolism of tryptophan, giving an essential group for constructing an NAD+ molecule [24]. NAD+ is known as a molecule that is involved in energy metabolism, substrate oxidation, and antioxidation [25,26,27]. In glycolysis, NAD+ is reduced to NADH when glyceraldehyde 3-phosphate is oxidized to bisphosphoglycerate. Accepting H-from NADH, pyruvate is reduced to lactate. Through this simple oxidization/reduction cycle of NAD+/NADH, one glucose molecule can produce two ATP and two lactate molecules independent of mitochondrial function or the respiration process. In this way, the Warburg effect would be observed specifically in most cancer cells. In normal cells, pyruvate is oxidized by pyruvate dehydrogenase [28] and converted to acetyl CoA, which can then be completely oxidized to CO2 and H2O through the TCA cycle. This reaction requires NAD+ as an acceptor of H-. Three enzymes, isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase, which are encoded by the IDH (IDH1, IDH2, IDH3A, IDH3B, and IDH3G), OGDH, and MDH (MDH1, MDH2, and MDH1B) genes, respectively, require NAD+ as a coenzyme for oxidization of each substrate in the TCA cycle [29]. As long as the mitochondrial electron transport chain functions, NADH will be immediately oxidized to NAD+ by complex I. Moreover, in β-oxidation of fatty acids, NAD+ is used as an acceptor of H- when 3-hydroxy-acyl-CoA dehydrogenase works. β-Hydroxybutyrate, which is one of the ketone bodies, is converted to acetoacetate. The reaction is catalyzed by β-hydroxybutyrate dehydrogenase (BDH), which is encoded on the BDH1 and BDH2 genes [24]. In the metabolism of nucleotide bases, NAD+ functions as a coenzyme for inosine 5′-phosphate (IMP) dehydrogenase (IMPDH) and dihydropyrimidine dehydrogenase (DPYD) [30], which are encoded by the IMPDH1/IMPDH2 and DPYD genes, respectively.
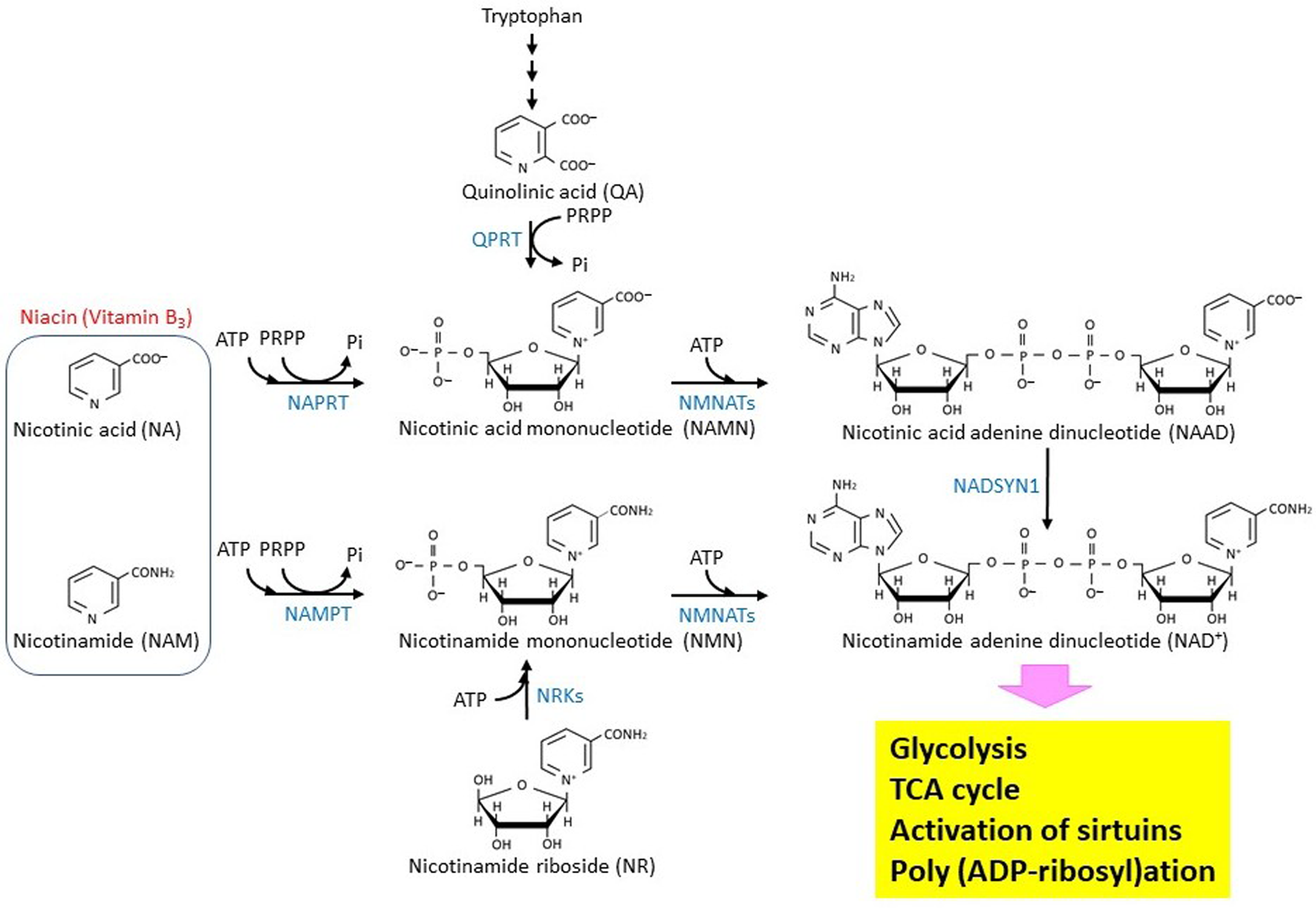
Figure 1. Biosynthesis and metabolism of NAD+ in eukaryotic cells. The biosynthetic pathway of the NAD+ molecule in eukaryotic cells is shown. The enzymes that catalyze the reactions are shown by blue characters. NMNATs are encoded by three different genes, NMNAT1 (GENE ID: 64802), NMNAT2 (GENE ID: 23057), and NMNAT3 (GENE ID: 349565). NRKs are encoded by two different genes, NMRK1 (GENE ID: 54981) and NMRK2 (GENE ID: 27231).
It should be noted that the NAD+ molecule is consumed for the synthesis of poly (ADP-ribose) (PAR) by PARP especially when DNA damage occurs [31]. The biological relevance of the NAD+ molecule in the protection of chromosomal DNAs has been reviewed and discussed previously [26,32]. A recent study showed that nuclear PAR can be used by NUDIX5 to supply ATP molecules, which are required for chromatin remodeling [33]. Moreover, NAD+-dependent deacetylases, sirtuin proteins that regulate both cancer generation and aging [34,35], possess histone deacetylase activity [36]. This observation suggests that sirtuins play roles in epigenetic regulation.
In summary, NAD+ is not only required for the oxidization process but also for DNA repair, chromatin remodeling, and epigenetic control. Importantly, these biological events that occur in the nucleus corelate with cancer generation/development and the aging process. Therefore, it is important to revisit the biosynthesis and metabolism of NAD+.
4. Biosynthesis and metabolism of NAD+
Figure 1 summarizes the biosynthesis and metabolism of the NAD+ molecule [26]. NAMPT is a nicotinamide phosphoribosyltransferase that catalyzes the first rate-limiting step of NAD+ synthesis from nicotinamide [37]. Therefore, its activity may modulate biologically important reactions such as the TCA cycle, poly (ADP-ribosyl)ation, and sirtuin-mediated de-acetylation that all depend on NAD+. The reaction product, nicotinamide mononucleotide (NMN), can be bound with ATP to produce NAD+ by nicotinamide nucleotide adenylyltransferases (NMNATs). Depletion of NMNAT2, which localizes in cytosol-facing Golgi apparatus, by siRNA introduction into cultured cells results in a decrease of both cytoplasmic and mitochondrial NAD+, suggesting that an NAD+ transport system is at work in mammalian cells [38]. NMN could be alternatively produced through nicotinamide riboside phosphorylation, which is catalyzed by nicotinamide riboside kinases. NAD+ can also be synthesized from nicotinic acid adenine dinucleotide (NAAD) by NAD synthetase (NADSYN1), and NAAD can be produced by de-phosphorylation of nicotinic acid adenine dinucleotide phosphate (NAADP) [39]. Moreover, it should be noted that PGC-1α, which is a pivotal regulator of mitochondrial biogenesis, up-regulates the de novo synthesis of NAD+ in mice [40].
Expression of the NAMPT gene is induced in glioblastoma cells and upregulates the E2F2/ID pathway [41]. A recent study revealed that the NAMPT/E2F2/SIRT1 axis promotes the proliferation of human melanoma cells [42]. Depletion of NAD+ by NAMPT inhibitor FK866 effectively leads to cell death in Ewing sarcoma [43]. A detailed analysis of metabolites revealed that in cancer cells, NAMPT inhibition suppresses both glycolysis and the TCA cycle [44]. These observations imply that NAMPT could be targeted in therapeutics for the treatment of some specific cancers, including gastric cancer and colorectal cancer, that overexpress the NAMPT (PBEF) gene [45,46,47]. The NAMPT protein was shown to form a complex with beta-actin and POTTE (POTE ankyrin domain family member E), and it might be responsible for the sensitivity to inhibitors such as FK866 [48]. Analyses of cancer genomes have revealed that the NAPRT, encoding nicotinate phosphoribosyltransferase, and NADSYN1 genes are amplified in many cancer types [49]. The NAPRT protein uses nicotinic acid as a substrate to produce nicotinic acid mononucleotide, suggesting that the Preiss-Handler (PH) pathway affects cancer generation. In addition, NAMPT gene transcription could be epigenetically controlled in non-PH-amplified cancer [49]. Another important NAD+ metabolism is the synthesis of S-adenosylhomocysteine (SAH) and a metabolically inert 1-methyl nicotinamide (1-MNA) by a nicotinamide N-methyltransferase (NNMT) that transfers a reactive methyl group from S-adenosylmethionine (AdoMet) to nicotinamide. AdoMet is generally known to be an essential donor of methyl group to histones, DNAs, and RNAs. Thus, NNMT-mediated AdoMet depletion affects gene expression [50]. Recently, it was shown that NNMT is a master metabolic regulator of cancer-associated fibroblasts [51]. In this regard, NAD+ metabolism could be linked with epigenetic regulation. Because nicotinamide, which is an acceptor of the methyl group, could play a role in depletion of AdoMet.
5. Possible roles of NAD+ in development of cancer and aging-related diseases
Not only the overproduction of NAD+ but also a decrease in its level can be problematic. NAD+ and its precursor nicotinamide can ameliorate metabolism or mitochondrial functions [52,53,54]. Repletion of NAD+ improves mitochondrial functions to prolong the life span of adult mouse stem cells [55]. Conversely, decreased concentrations of NAD+ will cause aging or aging-related diseases [56]. These observations are consistent with the concept that the NAD+ level correlates with mitohormesis [57] and that nutrient-sensing molecules could control aging [58].
In breast cancer cells, knockdown of the NDUFV1 subunit leads to an aberration in complex I to enhance aggressiveness or metastasis [59]. Therefore, upregulation of the mitochondrial NAD+ level will improve mitochondrial respiratory function or metabolism and that could suppress oncogenesis. Mitochondrial biogenesis is mainly regulated by PGC-1α, which drives NAD+ biosynthesis and thereby induces stress resistance [39]. PGC-1α suppresses the metastasis of melanoma by affecting the PGC-1α-ID2-TCF-integrin axis through transcriptional control [60]. A loss of mitochondria localizing protein CSB [61] can activate PARP1, which plays essential roles in DNA repair [62], thus consuming the NAD+ molecule. Therefore, dysregulation of the mitochondria or hyperactivation of the PARP enzyme will reduce the NAD+/NADH molecular ratio. Mutations on the IDH1 and IDH2 genes have been identified in human brain cancer cells [21,63,64]. In addition, mutation-introduced IDH2 could generate sarcomas [65]. Incomplete functioning of the IDH proteins might retard progression of the TCA cycle, thus leading to mitochondrial dysfunction. It should be noted that 5′-upstream regions of the TCA cycle enzyme-encoding genes, including IDH1, IDH3A, and IDH3B, contain duplicated GGAA motifs (Figure 2), which are recognized by various TFs, such as ETS family proteins [66]. Surveillance of the NCBI database (https://www.ncbi.nlm.nih.gov/gene/) indicated that the duplicated GGAA motifs are contained in the 5′-upstream regions of the TCA cycle enzyme-encoding genes, including PDH, MDH1, and MDH2 (Figure 2). Interestingly, the IDH3G, OGDH, and MDH1B gene promoters have no such elements, but the reactions that are catalyzed by the encoded enzymes require NAD+ as a coenzyme.
Metabolic dysfunction is thought to play a major role in the development of neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD) [67]. Thus, mitochondrial dysfunction is not only thought to be one of the causations of cancer but also of neurodegenerative disease [68]. For example, high levels of deletions in mtDNA have been identified in PD [69]. In addition, an NAD+-dependent deacetylase SIRT3, which localizes in mitochondria, may play a role in the development of neurodegenerative disorders, including AD, PD, and HD [70,71]. Caloric restriction (CR) attenuated the accumulation of amyloid-β peptide (Aβ) plaques in a transgenic mouse model of AD [72]. It has been reported that CR-induced SIRT1 reduced AD pathology [73]. Interestingly, SIRT2, which is a tubulin deacetylase, plays a role in accelerating Aβ aggregation [74]. In addition, SIRT6 can protect cells from Aβ-induced DNA damage [75]. Therefore, NAD+ is thought to play a role in the pathogenesis of AD and other neurodegenerative diseases by its effect on sirtuin proteins.
Figure 2. Human TCA cycle-associated protein-encoding genes that have duplicated GGAA motifs near the transcription start site.
6. NAD+-sensitive transcription control system
Epigenetic regulation, which is mainly driven by modifications of chromosomal DNAs and histone proteins [76], determines the chromatin compositions and structures [77]. Epigenetic alterations are frequently found in cancer, implying that “cancer is an epigenetic disease” [78]. The correlation between cellular metabolites and gene expression has been proposed as the RNA/enzyme/metabolite (REM) networking system [79]. Because the metabolites NAD+, AdoMet, and acetyl CoA are the substrates for poly (ADP-ribosyl)ation, methylation, and acetylation, respectively [80], they could be involved in epigenetic regulation.
The NAD+ molecule is the substrate of the PARP enzymes, which play pivotal roles in the DNA repair system [26,31,81]. The inhibition of PARP1 ameliorates mitochondrial metabolism through the activation of SIRT1 [82]. Conversely, over-activation of PARP1 can lead to mitochondrial dysfunction [83]. PARP1 gene expression was found to be negatively regulated when poly (ADP-ribose) glycohydrolase (PARG) siRNAs were introduced into HeLa S3 cells [84]. Therefore, degradation of the PAR macromolecule might be required for transcription of the PARP1 gene. It has been suggested that PARP1 is involved in the NAD+-sensitive transcription system [85]. Interestingly, duplicated GGAA motifs are commonly contained in the 5′-upstream regions of the human PARP1 and PARG genes [66,86], suggesting that both genes are regulated in an NAD+-dependent manner. The NAD+/NADH ratio may not only directly affect mitochondrial functions and DNA repair but also the transcription of DNA repair factor-encoding genes.
Presently, it has been known that trans-resveratrol (Rsv)-activated mitochondrial complex I upregulates the NAD+/NADH ratio [87], and that leads to the modification of chromatin-associating proteins [88] and modulation of the gene expression system [89]. The human TP53 gene promoter, which contains a duplicated GGAA motif, is up-regulated in HeLa S3 cells in response to Rsv [90]. Of note, the bidirectional BRCA1/NBR2 promoter region contains a duplicated GGAA motif and is regulated by a metabolic switch depending on the NAD+/NADH ratio, which could be elevated by CR-mimetic drugs [91]. The C-terminal binding protein (CtBP) [92,93] might have an essential role in this regulation as a metabolic sensor. Recently, it was shown that CtBP could work as an NAD(H)-sensitive transcriptional co-repressor that regulates NF-κB-driven pro-inflammatory transcription in macrophages and microglia [94]. Co-transfection of the CtBP1 or CtBP2 expression vector to CtBP1/CtBP2-knock down cells down-regulated the Luc activities of the NF-κB motif containing Luc reporter plasmid-transfected cells [94]. The NF-κB p65 (RELA) homodimer recognizes two symmetric sequences, 5′-GGAATTTCC-3′ and 5′-GGAATTCCC-3′, which are contained in the IL8 and COL7A1 enhancers, respectively [95]. Therefore, duplicated GGAA motifs might play a role in responding to CtBP that affects NF-κB, which can bind to the duplicated GGAA motifs. Moreover, the GABP TF (nuclear respiratory factor 2), which belongs to the ETS family and directly recognizes GGAA motifs, is required for mitochondrial biogenesis [96]. In summary, the duplicated GGAA (TTCC) motifs, which are very frequently contained in the 5′-upstream regions of a number of genes that encode DNA repair factors and TCA cycle-associating enzymes (Figure 2) [66,96], may be partly dependent on the level of NAD+ that affects activities of CtBP and GGAA motif-binding TFs. A recent study showed that mitochondrial LACTB could be a potent tumor suppressor [97]. Notably, the duplicated GGAA motifs are present in the 5′-upstream of the LACTB gene.
It is noteworthy that oxidative stress-responding TF Nrf2 recognizes antioxidant response element (ARE) in the promoter region of the NQO1 gene, encoding NAD(P)H:quinone oxidoreductase1 (NOQ1), and upregulates its expression [98]. Interestingly, ARE is contained in the promoter region of the ETS1 gene [99], which encodes TFs that bind to the GGAA motif-containing sequences [100]. Therefore, the Nrf2/ARE pathway may also contribute to regulating the expression of genes encoding proteins that play roles in the redox system or TCA cycle. Another important gene expression controlling system might be epigenetic alteration by sirtuin proteins, which are the NAD+-dependent deacetylases [101,102], and sirtuin deacetylate TFs such as FOXO, NF-κB, and STAT proteins [103,104]. Importantly, SIRT1 is involved in mitochondrial biogenesis depending on the metabolic state in muscle [105]. SIRT3, which localizes in mitochondria, can up-regulate functions of mitochondrial proteins, including TCA cycle enzymes [106]. SIRT1, SIRT2, and SIRT3 proteins are involved in the reactive oxygen species defense mechanism co-operatively regulating various TFs, including FOXO, HSF1, PGC-1α, and p53 [34]. Thus, NAD+ may modulate or fine-tune the transcriptional state of both DNA repair- and mitochondrial function-associated genes, which are required for cells to manage various stresses from, for example, DNA damage, nutrient conditions, and viral infections.
.../...
.
















