































.
Abstract
Skeletal muscle aging is associated with a significant loss of skeletal muscle strength and power (i.e., dynapenia), muscle mass and quality of life, a phenomenon known as sarcopenia. This condition affects nearly one-third of the older population and is one of the main factors leading to negative health outcomes in geriatric patients. Notwithstanding the exact mechanisms responsible for sarcopenia are not fully understood, mitochondria have emerged as one of the central regulators of sarcopenia. In fact, there is a wide consensus on the assumption that the loss of mitochondrial integrity in myocytes is the main factor leading to muscle degeneration. Mitochondria are also key players in senescence. It has been largely proven that the modulation of mitochondrial functions can induce the death of senescent cells and that removal of senescent cells improves musculoskeletal health, quality, and function. In this review, the crosstalk among mitochondria, cellular senescence, and sarcopenia will be discussed with the aim to elucidate the role that the musculoskeletal cellular senescence may play in the onset of sarcopenia through the mediation of mitochondria.
1. Introduction
Declines in skeletal muscle mass and function are among the most notable corollary of aging. Muscle mass reaches its peak between 30 and 40 years of age and starts declining thereafter [1]. Such a decline is considered a normal phenomenon during aging, but it can rapidly progress in physically inactive persons as well as in the setting of acute or chronic conditions. In particular, in people with an inactive lifestyle, the loss of muscle mass can reach 1% to 2% per year from age 50 to 60, and 3% to 5% per year at older ages [2]. As a result, an inactive person can lose from 30% to 50% of muscle mass between the ages of 40 and 80 years [2].
The term sarcopenia was coined by Rosenberg [3] from the Greek words “sarx” (flesh) and “penia” (poverty) to describe the loss of muscle mass with aging. This condition affects nearly one-third of the older population [2], and is one of the main factors leading to negative health outcomes in older adults [4]. Over the years, sarcopenia has been better defined through the inclusion of reduced muscle strength and/or function (i.e., dynapenia) [5,6,7] in the conceptual framework [8,9,10,11,12]. Indeed, sarcopenia has recently been formally recognized as a disease by means of a novel ICD-10-MC code [13].
Several cellular and molecular mechanisms are involved in the pathogenesis of age-related muscle wasting. During aging, a decrease in the number and size of muscle fibers occurs, a process defined by Lynch et al. [14] “remodeling of the motor unit in relation to age”. Fiber atrophy and demise, in turn, result from reduced protein synthesis [15] and impaired muscle regeneration [16]. Other factors that contribute to muscle loss in advanced age include neuromuscular junction dysfunction, reduced satellite cell number/function, decreased number of motor units [17], intramuscular adipose tissue infiltration [18], inflammation [19], insulin resistance [20], mitochondrial dysfunction [21], and oxidative stress [22].
Although the exact mechanisms responsible for the development and progression of sarcopenia are not fully understood, mitochondrial dysfunction has emerged as a central pathogenetic factor [23]. Indeed, mitochondria serve a number of vital functions within the cell, including energy production, regulation of intracellular calcium homeostasis, modulation of cell proliferation, and integration of apoptotic signaling. Hence, the preservation of well-functioning mitochondria is pivotal for maintaining cellular homeostasis [24]. When mitochondrial quality control fails, mitochondria lose their integrity and may cause muscle degeneration [25]. Notably, the accumulation of damaged mitochondria has shown to trigger motor neuron and muscle fiber death, highlighting their relevance in the development of sarcopenia [26].
Here, we summarize available evidence supporting mitochondrial dysfunction as a mechanism contributing to musculoskeletal aging and sarcopenia. We also illustrate the involvement of mitochondria in cellular senescence with the aim to highlight the relationship between musculoskeletal cellular senescence induced by mitochondrial dysfunction and the onset of sarcopenia.
2. Mitochondria and Aging
Several evolutionarily conserved biological pathways have been indicated as the main drivers of aging [27]. Such pathways, collectively known as hallmarks of aging, include mitochondrial dysfunction, genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient-sensing, cellular senescence, stem cell exhaustion, and altered intercellular communication [27]. Previous studies showed that mitochondrial dysfunction arising from the abnormal accumulation of mitochondrial DNA (mtDNA) induced the early appearance of several age-related phenotypes, including sarcopenia, in mice [28,29,30]. However, whether laboratory rodents genetically engineered to accumulate lower amounts of mtDNA mutations during aging are protected against sarcopenia remains to be proven.
Researchers have proposed several theories to explain the functional decline associated with age-related mitochondrial dysfunction (Table 1). In particular, the notion that a shift in redox status towards oxidation leading to progressive cellular decline has gained momentum [31]. In this regard, when the defense system is no longer able to cope with the enhanced rate of oxidant production, cellular, and subcellular environments become more susceptible to damage [31]. For this reason, older persons activate a compensatory mechanism of upregulation of antioxidant enzymes to counteract the increasing generation of reactive oxygen species (ROS) [32]. However, despite the overactivation of antioxidant pathways, a positive correlation between age and oxidative damage is well-established with advancing age [33]. The persistence of sustained oxidative stress in spite of upregulated antioxidant activity suggests that antioxidant defense mechanisms may be overwhelmed in advanced age [33].

Table 1. The Main Mitochondrial Theories of Aging.
MtDNA copy number also varies during aging and might be considered a biomarker that mirrors alterations within the aged human body [34,35,36]. A new quantitative, highly sensitive droplet digital PCR method has allowed observing a mild gradual age-related reduction of the mtDNA copies in stimulated peripheral blood mononuclear cells in a sample ranging in age between 23 and 113 years [37]. Interestingly, centenarians with lower levels of frailty showed a significantly higher number of mtDNA molecules and fewer mtDNA deletions compared with those with more severe frailty [37].
3. Mitochondria and the Skeletal Muscle
Skeletal muscle is highly represented in the human body and is responsible for intentional movements and postural maintenance [42,43]. It has also a crucial role in some less obvious processes, such as thermal regulation, nutritional balance, glucose uptake, and endocrine activity [44,45].
The skeletal muscle of humans and other mammals consists of different cell types, i.e., multinucleated myofibers (or myotubes) and satellite cells, all wrapped in the sarcolemma [46,47]. A single myofiber is a postmitotic highly differentiated cell, which contains numerous peripheral nuclei, myofilaments, the sarcoplasmic reticulum, and finally the mitochondria, which represent the actual machinery providing energy to the movement [48].
3.1. Mitochondria Localization in Skeletal Myofibers
Ultrastructural studies have identified in human and rodent muscles different subsets of mitochondria localized within the skeletal myotubes: subsarcolemmal, perinuclear, and intermyofibrillar mitochondria [49,50,51]. Every subtype of mitochondria has peculiar biochemical and proteomic specializations. In particular, subsarcolemmal mitochondria have a role in gene expression and resistance to ROS, whereas intermyofibrillar mitochondria are concerned with processes such as oxidative phosphorylation and the modulation of Ca2+ flux [52]. Para-vascular mitochondria are specific to vessels, but have also recently been found in skeletal myocytes [53]. The peculiarities of human skeletal muscle mitochondria are their dynamic behavior within the myofiber [54], and their connectivity and/or ramification in the muscle, that is determined by mtDNA and could be subject to change in response to aerobic oxidative metabolism [55,56].
Two different types of skeletal muscle fibers are known according to the different isoforms of structural proteins: the myosin heavy chain and the tropomyosin [57]. The most common are type I or slow-twitch myofibers, and type II, or fast-twitch myofibers. This last type is further divided into type II A and type II X [58,59]. Red muscles mainly consist of slow types II A and I fibers and rely mostly on aerobic oxidative metabolism, while white muscles are made up of fast type II B fibers and adopt glycolysis [60,61]. Interestingly, in anaerobic glycolytic fibers, mitochondria are associated with the sarcomere I-band, whereas in oxidative fibers, mitochondria are mainly accumulated in I-band and A-band [62]. Moreover, in fast-oxidative or fast intermediate myofibers which can be found in red muscles, all triads are associated with mitochondria causing Ca2+ release from sarcoplasmic reticulum and ATP production [63,64].
3.2. Mitochondria Dynamics in the Skeletal Muscle
Changing metabolic demands induce modifications in the shape and dynamics of mitochondria residing in skeletal muscle [65]. A constant balance is maintained between the amount of short and elongated fused mitochondria. This balance results in fusion and fission processes as well as on the activity of shaping proteins [66,67]. A major process regulating metabolic plasticity is mitophagy [68]. The latter involved an organelle-specific form of macro-autophagy that drives dysfunctional mitochondria towards degradation. The quality control system of mitophagy guarantees the maintenance of the cellular structure and mitochondrial integrity. Mitochondrial fission precedes mitophagy: elevated ROS levels and loss of mitochondrial membrane potential are two key events triggering mitophagy [69]. Mitophagy and fusion/fission events are dysregulated when muscle atrophy develops [70], during which muscle protein degradation is accentuated [71,72]. In such circumstances, mitochondria become shorter and fragmented, and mitophagy flux is upregulated [73,74].
4. Mitochondria and Sarcopenia
Mitochondria have been indicated as the main actors in the development of sarcopenia (Figure 1) [25,75].
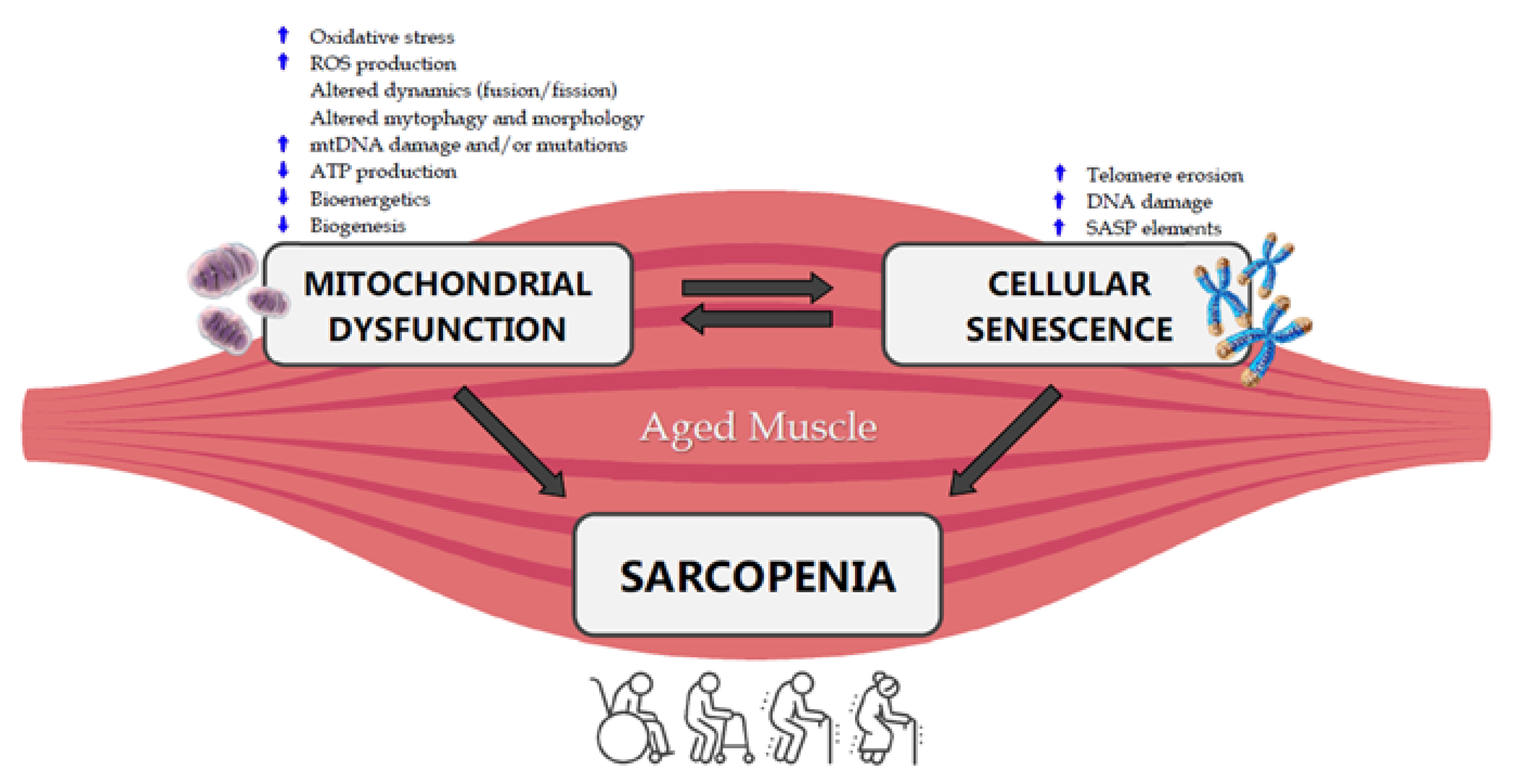
Figure 1. Crosstalk between mitochondrial dysfunction, cellular senescence and sarcopenia in the aged muscle. ROS: Reactive Oxygen Species, mtDNA: mitochondrial DNA, ATP: Adenosine Triphosphate, SASP: Senescence-Associated Secretory Phenotype.
.../...
.















