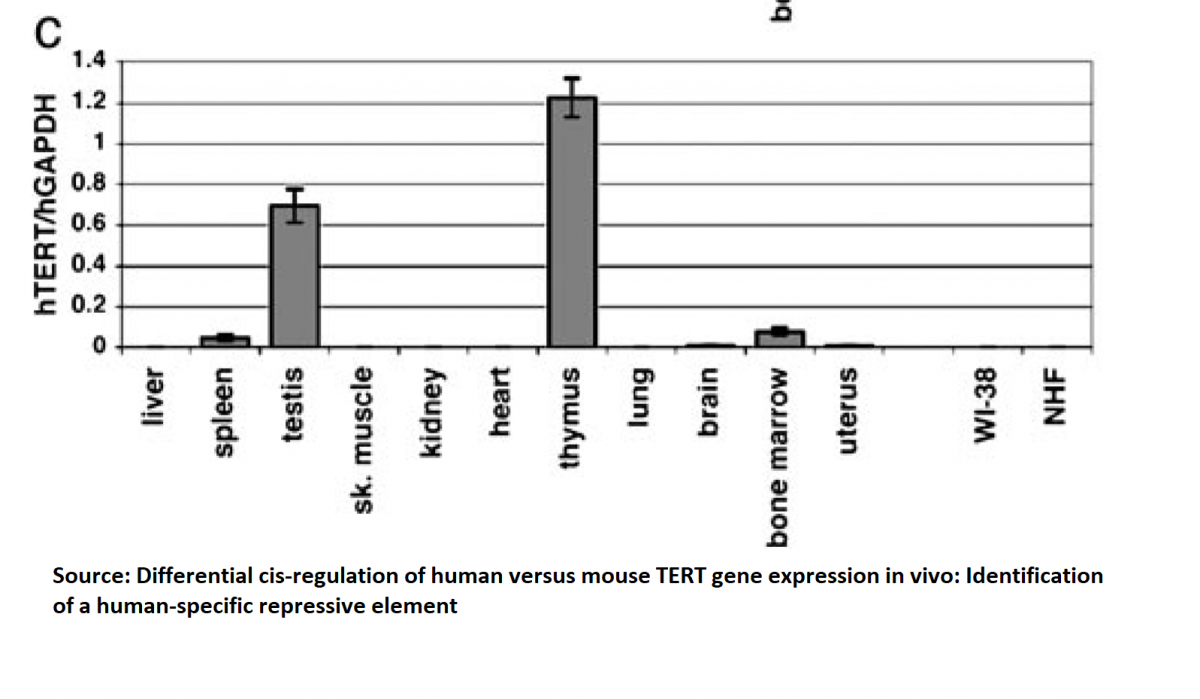
Telomere length and telomerase activity in T cells are biomarkers of high performing centenarians
It is generally recognized that the function of the immune system declines with increased age and one of the major immune changes is impaired T cell responses upon antigen presentation/stimulation. Some “high‐performing” centenarians (100+ years old) are remarkably successful in escaping, or largely postponing, major age‐re-lated diseases. However, the majority of centenarians (“low‐performing”) have experienced these pathologies and are forced to reside in long‐term nursing facilities. Previous studies have pooled all centenarians examining heterogeneous populations of resting/unstimulated peripheral blood mononuclear cells (PBMCs). T cells represent around 60% of PBMC and are in a quiescence state when unstimulated. However, upon stimulation, T cells rapidly divide and exhibit dramatic changes in gene expression. We have compared stimulated T‐cell responses
and identified a set of transcripts expressed in vitro that are dramatically different in high‐ vs.low performing centenarians. We have also identified several other measurements that are different between high‐ and low‐performing centenarians: (a) The amount of proliferation following in vitro stimulation is dramatically greater in high‐performing centenarians compared to 67‐ to 83‐year‐old controls and low‐performing centenarians; (b) telomere length is greater in the high‐performing centenarians; and telomerase activity following stimulation is greater in the high‐performing centenarians. In addition, we have validated a number of genes whose
expression is directly related to telomere length and these are potential fundamental biomarkers of aging that may influence the risk and progression of multiple aging conditions.
Source: DOI: 10.1111/acel.12859
An interesting paper from Jerry Shay and co. looking at telomerase activity and proliferative ability of T cells in centenarians compared to old (67-83yo) and young (~23-39yo) controls. Telomerase activity was elevated in centenarians compared to the old group, as (unsurprisingly) was proliferative ability of T cells (more telomerase meant they could make more T cells on stimulation, correlation between the two 0.88, p value <0.001).
T cells are different from most human cell types, in that they retain telomerase activity on differentiation, but only after stimulation with antigens.
Shay et al. also looked at gene expression assays and determined a clear difference between old and young donors, as well as a separation of the centenarians into two groups. The centenarian group that had gene expression more akin to young than old controls were found to be much
healthier than the centenarian group members with gene expression closer to the old controls. This 'health' was measured by better physical and cognitive performance, as well as fewer age related diseases. They were then designated High Performing centenarians, with the others labelled as Low Performing Centenarians.
They then went back and looked at the HP and LP centenarians and found that the high performers had longer telomeres and their 20% shortest telomeres were also significantly longer than the LP centenarians.
Finally they looked at what genes were expressed differently between old and young, as well as those genes that had similar expression between young and high performing (but not low performing) centenarians or the old control group. Interestingly, they found the genes that were maintained at a high level in young and HP centenarians were enriched for antigen response
pathways, and that genes that were kept at lower levels in young and HP centenarians (compared to old control and LP centenarians) included inflammatory response, IGF-1 expression and apoptosis signalling. They also found the differentially expressed genes (between old/LPC and young/HPC) were not randomly located but on certain chromosomes. They further found many were on chromosomes within 10MB of the telomere, as predicted by their Telomere Position effect over-long distance (TPE-OLD) model, whereby a shortening
telomere loses contact with genes it was formerly in contact with (via chromosome compaction and curling), changing the genes' expression.
It is not difficult for me to interpret this in the light of my Selfish Cell theory. Longer lived leukocytes with longer telomeres will acquire random methylation selecting for those less sensitive to antigen stimulation, preserving themselves, but harming the immune response. Those leukocytes that do respond, suffer from telomere shortening (even with activated telomerase), and in time are eliminated. Hence stronger signalling is required to get the needed response, including inflammatory and IGF-1 stress signals. Apoptosis is the result of the attrition of the limited number of leukocytes that can be made to respond.
From the other horn of aging, telomere shortening over time can result in deleterious gene expression changes (possibly via TPE-OLD), as well as a direct slowing of proliferation. As I said, T cells are a special case, as their ability to express telomerase is maintained on differentiation. So it is likley that methylation is primary here. Nevertheless, telomere length and telomerase expression still mediate most of the deleterious effects of aging in T cells. More research is required here.
The high performance of some centenarians cannot be attributed to better immune function alone. As we've seen in other studies, telomere length in T cells is correlated with telomere length in other tissues, therefore (Selfish Stem cells) in those tissues will also play their part in
aging. It is interesting that the HP centenarians had higher BMI than LP centenarians, who may have been suffering from tissue atrophy.
As for treatment, I'd expect exogenous telomerase treatment to counter the inability of aged T cells to replicate sufficiently to ward off infection. But I would also expect upregulation of demethylators (TETs) to have a beneficial effect on the immune system, via the upregulation of
telomerase that is secondary to a restored antigen response (assuming its promoters are subject to methylation), independent of telomerase activators. It will be interesting to see if this is confirmed by further work with AKG treatment of mice (or humans for that matter), to see if the previously reported improvements in frailty and inflammation are accompanied by improved immune function.
Edited by QuestforLife, 16 August 2021 - 08:23 PM.