Naringenin: super supplement?
1.Naringenin lengthens telomeres in mouse neural stem cells, reduces senescent cell markers and increases their proliferation (in vitro). It also enhances the learning and memory of aged mice (in vivo; injected).
Anti-aging effects of Ribes meyeri anthocyanins on neural stem cells and aging mice
Aging is associated with neurological impairment and cognitive decline. Flavonoids are very promising in anti-aging research in mouse models. Ribes meyeri anthocyanins are rich in abundant flavonoids, but their anti-aging biological activities remain unknown. In this study, we prepared an R. meyeri anthocyanin extract and analyzed its effects on neural stem cell (NSC) senescence in vivo and in vitro. We isolated mouse NSCs and used cell counting kit-8 (CCK-8), cell cycle, reactive oxygen species (ROS), and immunofluorescence methods to analyze the anti-aging effects of R. meyeri anthocyanins as well as naringenin (Nar), which metabolic analysis revealed as an important flavonoid in R. meyeri anthocyanins. RNA-sequencing (RNA-seq) and enzyme-linked immuno sorbent assay (ELISA) methods were also used to investigate Nar-specific mechanisms of anti-aging. After R. meyeri anthocyanin treatment, NSC proliferation accelerated, and NSCs had decreased senescence markers, and reduced P16ink4a expression. R. meyeri anthocyanin treatment also reversed age-dependent neuronal loss in vivo and in vitro. Nar blocked mNSC aging in vitro and improved spatial memory and cognitive abilities in aging mice through downregulation of plasma TNF-α protein. These findings suggest that R. meyeri anthocyanins increase NSC proliferation and improve neurogenesis with aging via Nar-induced reductions in TNF-α protein levels in vivo.
DOI: 10.18632/aging.103955
2. Naringenin (in doses of 1-50uM) acts through suppressing SMAD3, which is activated by TGF-B.
Smad3 Specific Inhibitor, Naringenin, Decreases the Expression of Extracellular Matrix Induced by TGF-"1 in Cultured Rat Hepatic Stellate Cells
Results. Naringenin reduced not only the accumulation of ECM, including collagen I!1 (Col I!1), fibronectin (FN), and plasminogen activator inhibitor-1 (PAI-1), but also the production of Smad3 induced by TGF-"1 in both mRNA and protein levels in a dose-dependent manner. Moreover,naringenin selectively inhibited the transcription of Smad3, but not other Smads involved in TGF-1 signaling pathways. Conclusion. Our data demonstrate that naringenin can exert antifibrogenic effects by directly or indirectly down-regulating Smad3 protein expression and phosphorylation through TGF-" signaling.
DOI: 10.1007/s11095-005-9043-5
3.SMAD3 is a negative regulator of telomerase activity. This is likely relevant to humans with already suppressed levels of telomerase, because the SMAD3 mediated suppression further reduced telomerase, even when its activity in rat cells had been impaired by a promoter mutation.
Role of Smad3 in the regulation of rat telomerase reverse transcriptase byTGFb
Telomerase is induced in certain pathological conditions such as cancer and tissue injuryand repair. This induction in fibroblasts from injured lung is repressed bytransforming growth factor b (TGFb) via yet unknown mechanisms. In this study, the role of Smad3 in the inhibition of telomerase reverse transcriptase (TERT) gene transcription byTGFb was investigated. The rat TERT (rTERT) gene promoter was cloned by PCR amplification and fused with a luciferase reporter gene. This construct was used to analyse regulation of promoter activity in fibroblasts isolated from bleomycin-injured lung with induced telomerase activity. The results showed that TGFb inhibited rTERT transcription while stimulating Smad3 expression. Interestingly, TGFb also inhibited the expression of c-myc. Cotransfection with a Smad3 expressing plasmid further repressed rTERT transcription and c-myc expression, while cotransfection with the corresponding antisense Smad3 construct had the opposite effect. Mutation of an E-box in the rTERT promoter suppressed its activity, which could be further reduced by TGFb treatment. In contrast, mutation at a Smad binding element enhanced promoter activity whose inhibition was impaired by TGFb treatment. Thus TGFb inhibition of rTERT gene expression was directly mediated by Smad3 via the Smad binding element, while c-myc appears to primarily regulate its constitutive or induced expression.
doi:10.1038/sj.onc.1209140
What is this I hear you cry? Those uM doses are quite unobtainable in a human! Normally with oral supplements I would agree, but…
4. Tens of uM can easily be obtained in human serum with doses of a few hundred mg. And it doesn’t build up either; even at the highest dose of 900mg Naringenin was completely cleared in 24 hours.
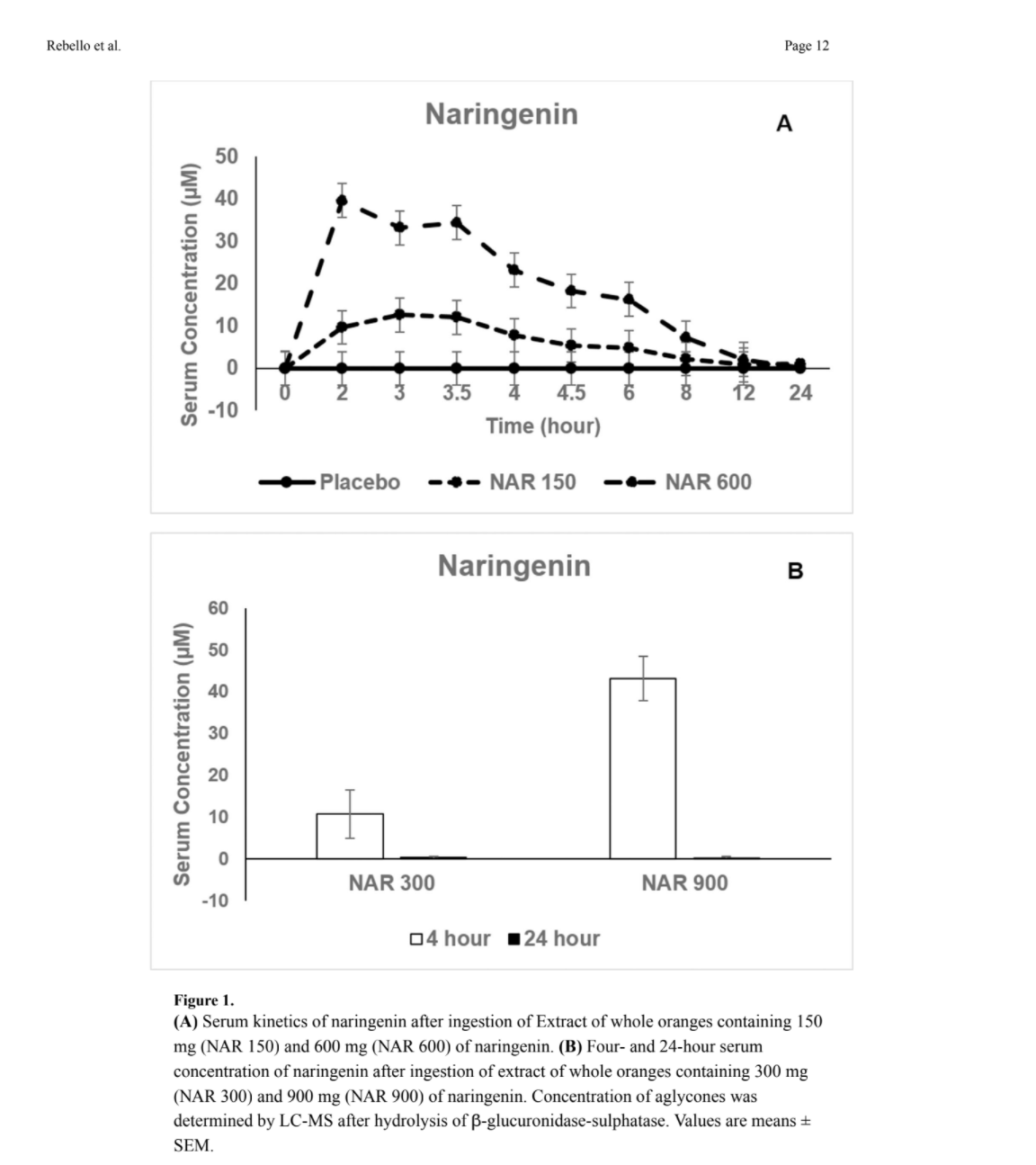
Safety and Pharmacokinetics of Naringenin: A Randomized, Controlled, Single Ascending Dose, Clinical Trial
Aims—This study evaluated the safety and pharmacokinetics of naringenin in healthy adults, consuming a whole orange (Citrus Sinensis) extract.
Methods—In a single ascending dose randomized crossover trial, 18 adults ingested 150mg (NAR150), 300mg (NAR300), 600mg (NAR600), and 900mg (NAR900) doses of naringenin or placebo. Each dose or placebo was followed by a wash-out period of at least one week. Blood safety markers were evaluated pre-dose and 24 hours post-dose. Adverse events were recorded. Serum naringenin concentrations were measured before and over 24 hours following ingestion of placebo, NAR150, and NAR600. Four and 24-hour serum measurements were obtained after
placebo, NAR300, and NAR900 ingestion. Data were analyzed using a mixed effects linear model.
Results—There were no relevant adverse events or changes in blood safety markers following ingestion of all naringenin doses. The pharmacokinetic parameters were: Maximal concentration:
15.76±7.88μM (NAR150) and 48.45±7.88μM (NAR600); Time to peak: 3.17±0.74h (NAR150) and 2.41±0.74h (NAR600); Area under the 24-hour concentration-time curve: 67.61±24.36μM×h (NAR150) and 199.06±24.36μM×h (NAR600); and Apparent oral clearance: 10.21±2.34L/h (NAR150) and 13.70±2.34L/h (NAR 600). Naringenin half-life was 3.0h (NAR150) and 2.65h (NAR600). After NAR300 ingestion, serum concentrations were 10.67±5.74μM (4h) and 0.35±0.30μM (24h). After NAR900 ingestion, serum concentrations were 43.11±5.26μM (4h) and 0.24±0.30μM (24h).
Conclusions—Ingestion of 150 to 900mg doses of naringenin is safe in healthy adults, and serum concentrations are proportional to the dose administered. Since naringenin (8μM) is
effective in primary human adipocytes, ingestion of 300mg naringenin twice/day will likely elicit a physiologic effect.
doi:10.1111/dom.13868.
I’ve attached a figure showing serum concentration.
What other benefits might we obtain from narigenin?
5. You might lose weight.
Naringenin Promotes Thermogenic Gene Expression in Human White Adipose Tissue
Objective—Naringenin, a citrus flavonoid, prevents diet-induced weight gain and improves glucose and lipid metabolism in rodents. There is evidence that naringenin activates brown fat and increases energy expenditure in mice, but little is known about its effects in humans. Our goal was to examine the effects of naringenin on energy expenditure in adipose tissue.
Methods—Human white adipocyte cultures (hADSC), and subcutaneous abdominal adipose tissue (pWAT) were treated with naringenin for 7–14 days. Expression (qRT-PCR, immunoblotting) of candidate genes involved in thermogenesis and glucose metabolism was measured. Oxygen consumption rate (OCR) was measured in hADSC using a Seahorse Flux analyzer.
Results—In hADSC, naringenin increased expression of the genes associated with thermogenesis and fat oxidation including uncoupling protein 1, adipose triglyceride lipase, and key factors associated with insulin sensitivity including glucose transporter 4, adiponectin, and carbohydrate response element binding protein (p<0.01). Similar responses were observed in pWAT. Basal, ATP-linked, maximal, and reserve OCR increased in the naringenin-treated hADSC (p<0.01).
Conclusions—Naringenin increases energy expenditure in hADSC and stimulates expression of key enzymes involved in thermogenesis and insulin sensitivity in hADSC and pWAT. Naringenin may promote conversion of human white adipose tissue to a brown/beige phenotype.
doi:10.1002/oby.22352.
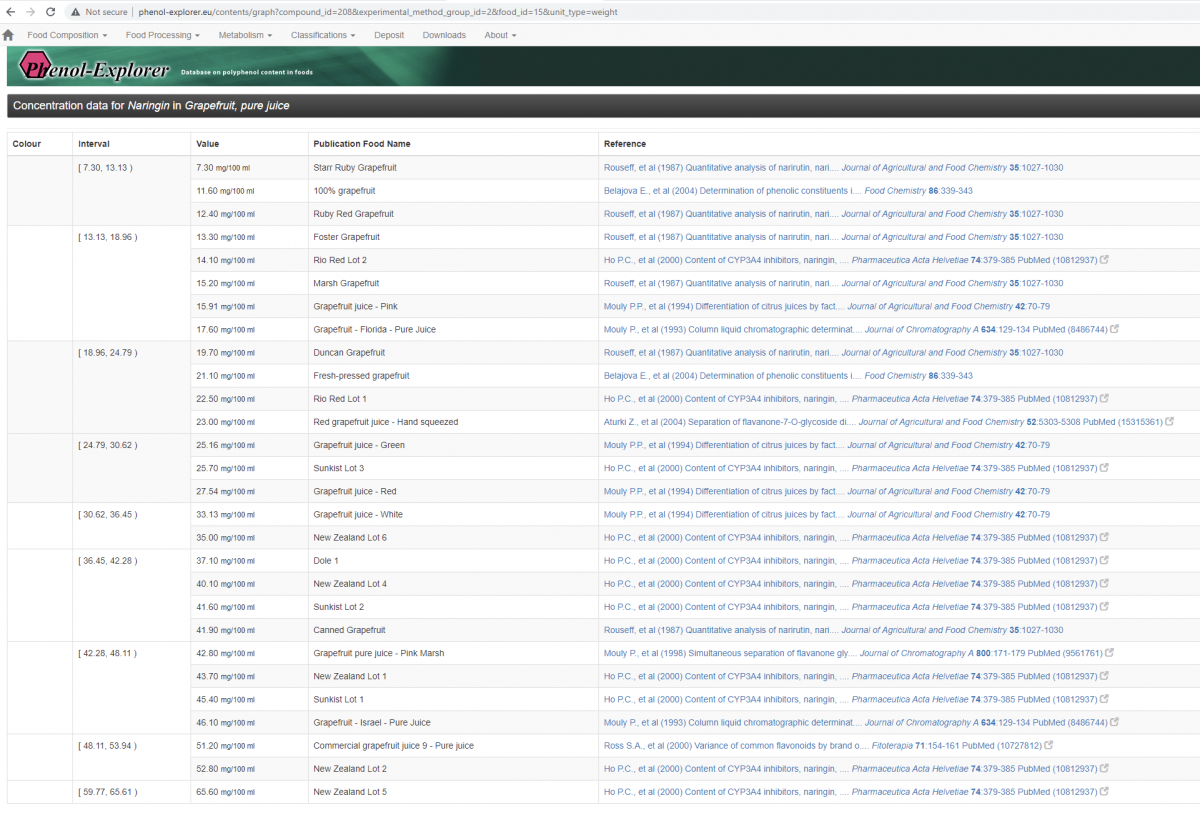
6.Conclusions: Narigenin looks like an easy win for telomeres and health in general; it is super bioavailable, is cleared quickly enough not to worry about taking breaks. How do we supplement? I attach a figure showing you can get a very decent dose of naringin from grapefruit juice. You can also buy near 100% naringin capsules if you desire. Note this is naringin not naringenin, but the former is rapidly converted into the later in the human gut by naringinase. Is there any downside? Grapefruit juice is said to inhibit the CYP3a4 enzyme, responsible for breaking down numerous pharmaceuticals (like rapamycin) and coffee, for example. So take grapefruit juice with care if you are on medication. But it is controversial what component of grapefruit is responsible for this, as a study showed quite weak inhibition of CYP3a4 by naringenin or naringin.