.
O P E N A C C E S S S O U R C E : Journal of Cell Biology
Mitochondria, long viewed solely in the context of bioenergetics, are increasingly emerging as critical hubs for intracellular signaling. Due to their bacterial origin, mitochondria possess their own genome and carry unique lipid components that endow these organelles with specialized properties to help orchestrate multiple signaling cascades. Mitochondrial signaling modulates diverse pathways ranging from metabolism to redox homeostasis to cell fate determination. Here, we review recent progress in our understanding of how mitochondria serve as intracellular signaling platforms with a particular emphasis on lipid-mediated signaling, innate immune activation, and retrograde signaling. We further discuss how these signaling properties might potentially be exploited to develop new therapeutic strategies for a range of age-related conditions.
For those of us who survived the emotional trauma known as junior high school in America, the strategy for adolescent success appeared to be the unique ability to both blend in and stand out. While we will leave it to social scientists to describe the psychological ramifications of such approaches, we would argue that there is an important hidden biological lesson here as well. Mitochondria, present in hundreds to thousands of seemingly identical copies per cell, certainly can blend in. In doing so, they provide the cell with its bioenergetics requirements, supplying the chemical energy required to power essentially all cellular processes. Yet, to view these structures as indistinguishable and amorphous, organelles would be shortsighted. Endowed with a unique evolutionary history, mitochondria have retained a distinct set of lipid components, as well as their own genome, that under specific stress conditions, enables these organelles to also stand out. This singularity makes mitochondria uniquely situated to act as a signaling platform. Here, we review the evidence for how mitochondria participate in a wide range of cell fate decisions that exploit the unique qualities and properties of this organelle. In particular, we will focus on how specific unique mitochondrial lipids can modulate signaling events, how mitochondria participate in innate immune signaling, and how mitochondria can signal back to the nucleus to alter both transcription and the epigenome. These topics cover the multifaceted stress responses used by mitochondria, from signaling reactions within mitochondria, to the surface of mitochondria, to those pathways involving the nucleus and the extracellular environment. Finally, we will describe the initial foray into leveraging these observations to develop new classes of therapies that may have the potential to treat a wide array of diseases, especially age-related diseases that are closely linked to mitochondrial dysfunction.
Mitochondrial lipid signaling
Involved in all aspects of cell biology and physiology, lipids are the major components of all cellular membranes, delineating the boundary between cells and subcellular organelles and supporting intercellular and intracellular communication. The most abundant lipid in mammalian cells is phosphatidylcholine (PC), whereas the amounts of other lipids such as phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), phosphatidic acid (PA), cholesterol, and sphingolipids vary depending on the type of cell and subcellular organelle. As an α-proteobacteria-derived organelle, mitochondria exhibit distinct lipid composition in both their inner and outer membranes, which establishes a biochemical basis for mitochondrial signaling. Mitochondrial lipid synthesis and transport have been the subject of several excellent recent reviews (Horvath and Daum, 2013; Tatsuta and Langer, 2017; Tatsuta et al., 2014), and as such, here, we will focus instead on mitochondrial signaling and metabolism regulated by two unique lipids, cardiolipin and PE (Fig. 1).
Cardiolipin-mediated signaling
Cardiolipin comprises nearly 20% of the inner mitochondrial membrane (IMM) and a much smaller fraction (up to 3%) of the outer mitochondrial membrane (OMM), but it is not found in any other subcellular organelle (de Kroon et al., 1997; Gebert et al., 2009). Structurally, cardiolipin is composed of a dimeric phosphatidylglycerol lipid, with two PA molecules connected with a glycerol backbone. This cone-shaped structure makes cardiolipin ideal for the highly curved IMM, the structure of which is dramatically disrupted when cardiolipin is depleted (Dudek, 2017). Cardiolipin is also well known for its integration into all respiratory chain complexes in the IMM and several translocase of outer membrane protein complexes (Dudek, 2017; Schlame and Greenberg, 2017; Xu et al., 2006). In humans, cardiolipin is synthesized at the IMM by cardiolipin synthase, followed by acyl chain remodeling leading to the formation of mature cardiolipin with polyunsaturated fatty acyl chains (Schlame and Greenberg, 2017; Tatsuta et al., 2014). Perturbing cardiolipin synthesis or remodeling causes mitochondrial dysfunction including defective oxidative phosphorylation and severe oxidative stress, as seen in Barth syndrome, a devastating X-linked pediatric disease (Schlame and Greenberg, 2017). In mice, deletion of cardiolipin synthase is embryonic lethal, whereas neuronal specific depletion causes disrupted mitochondrial crista structure and respiration, altered mitochondrial calcium handling, and neurodegeneration (Kasahara et al., 2020), indicating an essential role for cardiolipin in mitochondrial structure and function.
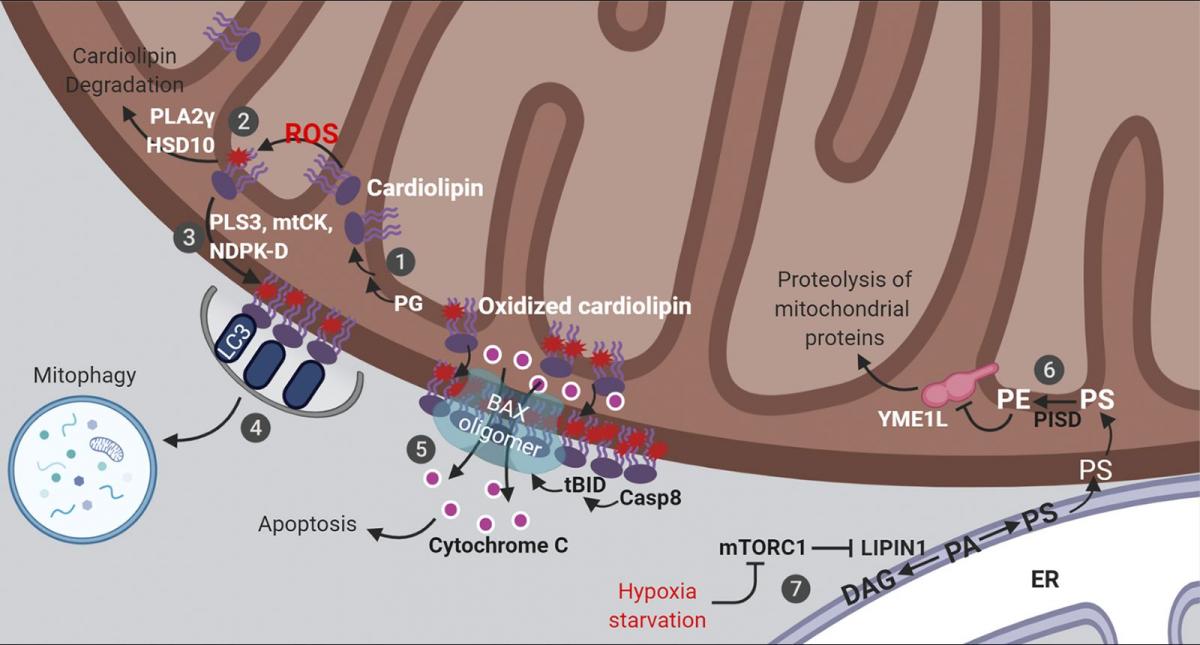
Cardiolipin functions as a reactive oxygen species (ROS) scavenger that protects cells from oxidative stress via cardiolipin oxidation and degradation (Fig. 1). ROS in animal cells is largely produced by the respiratory chain complexes which are assembled in the cardiolipin-rich IMM. The proximity of cardiolipin to the source of ROS and the presence of polyunsaturated fatty acyl chains in cardiolipin makes it an ideal sensor and scavenger of oxygen radicals. Oxidized cardiolipin is toxic (Paradies et al., 2001, 2002) and needs to be quickly degraded by enzymes including phospholipase A2γ (PLA2γ; Liu et al., 2017; Tyurina et al., 2014) and 17-β-hydroxysteroid dehydrogenase 10 (HSD10 [also known as amyloid β-peptide-binding alcohol dehydrogenase]; Boynton and Shimkets, 2015). PLA2γ, which can directly hydrolyze cardiolipin, appears to be the major enzyme for the degradation of oxidized cardiolipin, since either genetic depletion or pharmacological inhibition of PLA2γ results in robust accumulation of oxidized cardiolipin upon mitochondrial stress (Liu et al., 2017; Tyurina et al., 2014). HSD10 uses a different mechanism by in fact further oxidizing cardiolipin, causing subsequent spontaneous breakdown of the oxidized product into diacylglycerol, dihydroxyzcetone, and orthophosphate (Boynton and Shimkets, 2015). Thus, to ensure efficient removal of oxidized cardiolipin, the protein levels and enzymatic activity of PLA2γ and HSD10 must be tightly regulated. Indeed, increased levels of HSD10 have been observed in patients with neurological conditions, including multiple sclerosis and Alzheimer’s disease (Krištofiková et al., 2009). In that regard, amyloid β peptide, which is mechanistically linked to Alzheimer’s disease, directly binds and inhibits the enzymatic activity of HSD10 (Yan et al., 1997; He et al., 1998). Moreover, in a rotenone-induced rat model of Parkinson's disease, inhibiting PLA2γ activity causes increased oxidative stress, more mitochondrial lipid oxidation, augmented apoptosis, and a subsequent exacerbation of Parkinson’s disease symptoms (Chao et al., 2018). From a signaling viewpoint, metabolism of oxidized cardiolipin by PLA2γ and HSD10 generates a large group of second messengers, including oxidized fatty acid lipids and diacylglycerol (Boynton and Shimkets, 2015; Liu et al., 2017; Tyurina et al., 2014), which may be involved in mediating the cellular response to mitochondrial stress.
Cardiolipin externalization from the IMM to the OMM is another important lipid reorganization event that often occurs in response to various conditions involving moderate levels of mitochondrial damage (Fig. 1). In healthy cells, cardiolipin is maintained at very low levels on the OMM and almost exclusively accumulates within the inner membrane (∼96.5 mol%; Kagan et al., 2016). In contrast, with mitochondrial damage or membrane depolarization, cardiolipin moves to the OMM. Three proteins have been reported to transport cardiolipin from the IMM to the OMM: phospholipid scramblase-3 (Liu et al., 2003), mitochondrial creatine kinase, and nucleoside diphosphate kinase D (Epand et al., 2007; Kagan et al., 2016; Schlattner et al., 2013). Although it is not clear whether these proteins work sequentially or in parallel, individual depletion of either phospholipid scramblase-3 or nucleoside diphosphate kinase D substantially reduces cardiolipin relocalization to the OMM (Chu et al., 2013; Kagan et al., 2016; Liu et al., 2008; Schlattner et al., 2013). Direct lipid transport through membrane contact sites between subcellular organelles have been described (Cockcroft and Raghu, 2018; Scorrano et al., 2019; Wu et al., 2018), but it is not clear exactly how these three cardiolipin transporters act, and whether they localize to IMM–OMM contact sites (Horvath et al., 2015).

Figure 1. Lipid signaling in mitochondria. Mitochondrial cardiolipin and PE orchestrate multiple aspects of mitochondrial signaling and bioenergetics. These are summarized here diagrammatically. (1) Cardiolipin is synthesized from phosphatidylglycerol (PG) and remodeled at the IMM. (2) Cardiolipin acts as a ROS scavenger via oxidation and redox-mediated degradation. (3) Moderate levels of mitochondrial stress stimulates cardiolipin externalization. (4) Exposed cardiolipin can be recognized by LC3, stimulating mitophagic clearance. (5) Severe stress triggers cardiolipin-mediated cytochrome c release and apoptosis. (6) PE on the IMM is directly produced by the enzyme PISD, using PS synthesized on the ER and transported to mitochondria. (7) Stresses that inhibit mTORC1 rewires mitochondrial metabolism via a lipid signaling cascade, reducing PE levels on the IMM, leading to augmented YME1L activity, increased proteolysis and reduced mitochondrial biogenesis. DAG, diacylglycerol; mtCK, mitochondrial creatine kinase; tBID, truncated BH3-interacting domain death agonist. See text for additional details.
Externalized, oxidized cardiolipin at the OMM establishes a signaling platform to orchestrate cellular response to mitochondrial damage that can include apoptosis or mitophagy (Fig. 1). In mitochondrial-dependent apoptosis, the recruitment and activation of the apoptotic initiator protease caspase-8, its substrate BH3-interacting domain death agonist (BID), and the downstream effector BCL-2–associated X (BAX) are all dependent on the presence of cardiolipin in the OMM (Gonzalvez et al., 2008; Kuwana et al., 2002; Lutter et al., 2000). The interactions among truncated BID (tBID), BAX, and cardiolipin drive the formation of supramolecular complexes containing BAX oligomers on the OMM surface that mediate membrane permeabilization and cytochrome c release (Kuwana et al., 2002; Lai et al., 2019; Lutter et al., 2000). The oxidation of cardiolipin reduces its interaction with cytochrome c, which further accelerates cytochrome c release to initiate apoptosis. In mitophagy-promoting conditions, increased cardiolipin at the OMM functions as an “eat-me” signal, recognized by the autophagy marker protein microtubule-associated-protein-1 light chain 3 (LC3) allowing for autophagic capture of the damaged mitochondria (Chu et al., 2013). As such, interfering with cardiolipin synthesis or maturation or its IMM-to-OMM transport potently affects mitophagic and apoptotic signaling (Chu et al., 2013; Garcia Fernandez et al., 2000; Hsu et al., 2015; Kagan et al., 2016).
Cardiolipin has been shown to interact with a litany of proteins linked to both protein aggregation and neurodegenerative diseases. Proteins such as α-synuclein, amyloid β, and Tau, known to form aggregates in neurodegenerative diseases, have all been reported to bind to cardiolipin-containing membranes (Ahyayauch et al., 2012; Camilleri et al., 2013, 2020; Nakamura et al., 2011; Robotta et al., 2014). Such binding is believed to reduce cardiolipin levels and/or interfere with cardiolipin function in mitochondrial respiration and/or ROS sensing. However, cardiolipin may play a role in resolving these protein aggregates, including amyloid β (Ordóñez-Gutiérrez et al., 2015) and α-synuclein fibrils (Ryan et al., 2018). Thus, further work is needed to more precisely define the physiological relationship between cardiolipin and protein aggregates in neurodegeneration.
Sphingolipids, usually maintained at very low levels in mitochondria compared with other organelles (the Golgi complex, endolysosomes, and the plasma membrane), have been increasingly implicated in age-dependent mitochondrial dysfunction. Ceramide, which is at the center of sphingolipid metabolism, accumulates in response to almost all types of cellular stress, including chemotherapeutic stress, ionizing and ultraviolet radiation, serum starvation, injury, and infection (Ogretmen, 2018). Like cardiolipin, ceramide can also induce apoptosis by triggering BAX-dependent mitochondrial outer membrane permeabilization (Birbes et al., 2001; Ganesan et al., 2010). Mechanistically, this may involve the direct targeting of voltage-dependent anion channel 2 (VDAC2) by ceramide (Dadsena et al., 2019). In addition, during the initial phase of mitochondrial-mediated apoptosis, ceramide is further processed into two lipid messengers, sphingosine-1-phosphate and hexadecenal, which specifically activate Bcl-2 homologous antagonist killer (BAK) and BAX, respectively, for mitochondrial membrane permeabilization (Chipuk et al., 2012). Moreover, ceramide may also contribute to mitophagy, as the lethal autophagy induced by a natural bioactive sphingolipid, N-stearoyl-D-erythro-sphingosine (C18-ceramide), is dependent on the direct interaction between C18-ceramide and LC3 (Sentelle et al., 2012). As might be expected, crosstalk between cardiolipin and ceramide exists. For instance, cardiolipin has been shown to inhibit ceramide synthesis and promote its cleavage by ceramidase (El Bawab et al., 2001; Kim et al., 2016; Okino et al., 2003). On the other hand, manipulation of ceramide modulates in vivo cardiolipin levels (Babenko and Storozhenko, 2017), which is consistent with observations of an age-related accumulation of ceramide and a corresponding decline in cardiolipin (Helmy et al., 2003; Monette et al., 2011; Petrosillo et al., 2008; Sen et al., 2007). In addition, alterations in these sphingolipid-dependent pathways have been increasingly linked to conditions such as Alzheimer’s disease (Pera et al., 2017).
Mitochondrial PE signaling
The IMM also contains the highest molar ratio of PE compared with other subcellular membranes, which is consistent with its bacterial origin, as PE is quite abundant in many bacteria (Kaval and Garsin, 2018; López-Lara and Geiger, 2017). This is mainly achieved by direct synthesis of PE from PS at the IMM (Fig. 1). Most phospholipids, including PC, PS, and PI, are synthesized in the ER and then transported to other organelles (Tatsuta and Langer, 2017). PS synthesized in the ER is transported to the mitochondrial outer membrane at ER–mitochondrial contact sites. PS is then further transferred from the mitochondrial outer to the inner membrane after which it is converted by PS decarboxylase (PISD) into PE (Schuiki and Daum, 2009). PISD is an enzyme conserved from bacteria to humans that is critical for PE synthesis at the IMM, malfunction of which causes mitochondrial fragmentation and embryonic lethality in mice (Hartmann et al., 2016; Steenbergen et al., 2005), indicating that direct PE synthesis at the IMM cannot be compensated by other pathways.
PE synthesis at the IMM is dynamically controlled to balance cell growth and mitochondrial biogenesis. One key protein regulated by PISD-produced PE at the IMM is YME1L, a transmembrane ATP-dependent protease essential for mitochondrial proteostasis and dynamics, apoptotic resistance, and cell proliferation (Anand et al., 2014; Stiburek et al., 2012). Normal levels of PE suppresses the proteolytic activity of YME1L (Fig. 1). However, stresses such as hypoxia or nutrient deprivation induce a lipid signaling cascade upon inactivation of mechanistic target of rapamycin complex 1 (mTORC1) to reduce PE levels at the IMM, leading to activation of YME1L-mediated proteolysis of mitochondrial proteins and subsequent inhibition of mitochondrial biogenesis (MacVicar et al., 2019). This involves activation of LIPIN1, a substrate of mTORC1 that is phosphorylated and hence inhibited under basal conditions (e.g., available oxygen and nutrients). Inactivation of mTORC1 leads to dephosphorylation and activation of LIPIN1 that reduces the level of PA, resulting in a decline in the subsequent synthesis of PS, a substate of PE (MacVicar et al., 2019). Consistent with a key role for PE in suppressing YME1L, blocking PS transfer from the mitochondrial outer to inner membrane (hence reducing PE levels) activates YME1L (MacVicar et al., 2019). Thus, signals that modulate lipid transport and/or synthesis pathways can rewire mitochondrial metabolism in response to physiological changes. Interestingly, the proteolytic degradation of YME1L, as well as another mitochondrial protease, OMA1, is regulated by various cellular insults that perturb mitochondrial functions (Baker et al., 2014; Rainbolt et al., 2016). It would therefore be important to investigate whether this phenomenon is also modulated by changes in mitochondrial lipids.
While mitochondrial cardiolipin and PE have been extensively studied, other potential lipid signaling pathways are not yet well characterized. Although the mitochondrial outer membrane is very weakly charged, dynamic turnover of certain negatively charged phosphoinositides has been reported. For example, a low level of phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] can be detected on mitochondria (Watt et al., 2002). Removal or masking this mitochondrial pool of PI(4,5)P2 leads to fragmentation and autophagic targeting of mitochondria (Rosivatz and Woscholski, 2011). However, the downstream effectors for these and related lipid signals are largely undefined.
In summary, the double-membrane structure of mitochondria and its unique lipid composition establish an unusual membrane environment that are essential for maintaining mitochondrial activity and homeostasis. In particular, mitochondrial lipid signaling, mediated through cardiolipin and PE, provides an immediate capacity to both sense and respond to mitochondrial stress. When this stress goes beyond the handling capacity of local lipid signaling, more mitochondrial damage is induced. This often leads to the activation of the next level of response, both on the surface mitochondria and in the cytosol, as we discuss below.
Mitochondrial innate immune signaling
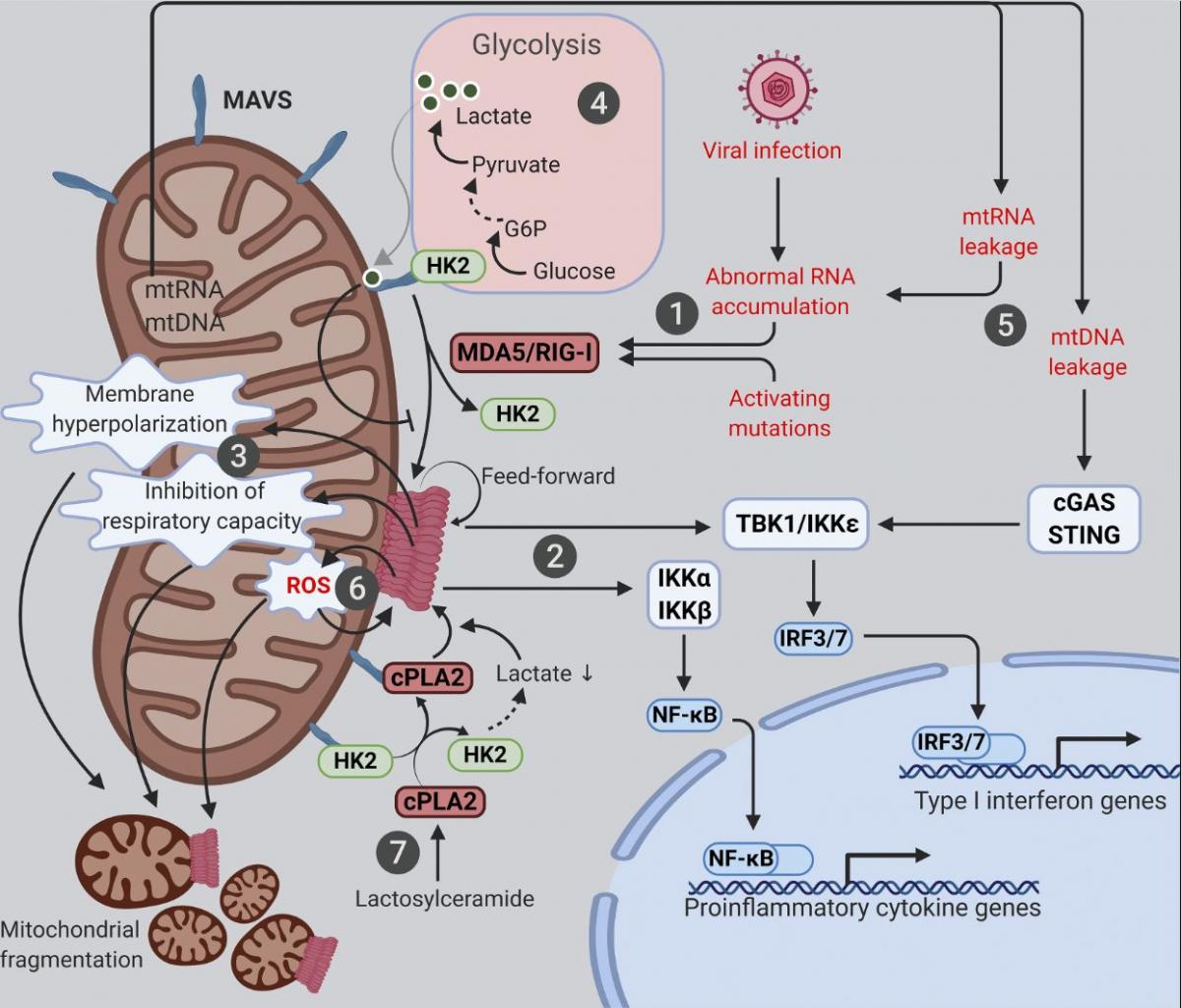
Mitochondria are linked to various innate immune signaling pathways, with the best characterized being the cytosolic RNA-sensing pathway for which mitochondria function as an essential platform (Fig. 2). While endolysosomal microbial RNAs are sensed by Toll-like receptors (specifically Toll-like receptors 3/7/8), those in the cytosol are directly captured by cytosolic RNA sensors known as retinoic acid–inducible gene I (RIG-I)-like receptors (RLRs; Tan et al., 2018). These include RIG-I (Yoneyama et al., 2004), melanoma differentiation-associated gene 5 (MDA5; Kang et al., 2002), and laboratory of genetics and physiology 2 (Saito et al., 2007), the last of which has no known intrinsic signaling capacity. Once bound to RNA ligands, both RIG-I and MDA5 form oligomers that directly bind and activate mitochondrial antiviral-signaling protein (MAVS [also known as IPS-1, VISA, and CARDIF]), a signaling scaffold with a C-terminal transmembrane domain anchored on the OMM (Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005). Activated MAVS forms aggregates that recruit signaling molecules (e.g., inhibitor of nuclear factor-κB kinase [IKK] and TANK-binding kinase [TBK1]), leading to the activation and nuclear translocation of NF-kB and IRF3/7, initiating transcriptional up-regulation of proinflammatory cytokines and type I IFNs (Liu et al., 2013). One key feature of MAVS signaling are its feedforward properties, whereby a small amount of MAVS aggregates can induce the formation of large MAVS aggregates by directly recruiting and activating other MAVS molecules on the mitochondria (Cai et al., 2014; Hou et al., 2011; Xu et al., 2015). The anchorage of MAVS on the mitochondrial surface is critical for its function, as removal of the MAVS transmembrane domain by a hepatitis C virus–encoded protease abrogates its signaling capacity (Li et al., 2005a; Li et al., 2005b; Meylan et al., 2005). Evidence suggests that MAVS may also localize to peroxisomes and mitochondrial-associated membranes (Dixit et al., 2010; Horner et al., 2011).
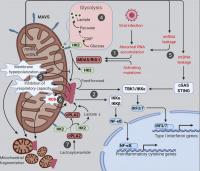
Figure 2. Mitochondria at the crossroads of innate immune signaling and metabolism. Mitochondria are platforms for MAVS innate immune signaling that modulates mitochondrial function and integrates immunity with cellular metabolism. The various known intersections between MAVS and mitochondrial function are diagramed. (1) Cytosolic RNAs perceived as foreign activate MAVS aggregation via the RNA sensors MDA5 and RIG-I. (2) Mitochondrial MAVS aggregates activate signaling cascades, leading to transcriptional induction of type I IFNs and proinflammatory cytokines. (3) MAVS aggregation causes marked alterations in a range of mitochondrial functions. (4) MAVS signaling inhibits glycolysis by releasing HK2 from mitochondria; lactate, a metabolite of anaerobic glycolysis, in turn suppresses MAVS aggregation via direct binding. (5) Mitochondrial leakage of RNA and DNA causes inflammation through MAVS and STING pathways, respectively. (6) Feedforward mechanism exists between MAVS aggregation and mitochondrial ROS. (7) Deregulated sphingolipid metabolism activates MAVS signaling independently of RNA ligands. cPLA2, cytosolic phospholipase A2; G6P, glucose 6-phosphate; HK2, hexokinase 2.
Cross-talk between MAVS signaling and cellular metabolism
There is extensive communication between MAVS signaling and mitochondrial metabolism (Fig. 2). It is well established that MAVS is C-terminally anchored on the OMM and that the formation of large MAVS aggregates causes substantial changes to mitochondrial morphology (Cai et al., 2014; Hou et al., 2011; Xu et al., 2015). Even endogenous levels of MAVS aggregates are sufficient to drive apoptosis, which is dependent on the MAVS transmembrane domain, likely linked to MAVS-driven mitochondrial fragmentation (Hwang et al., 2019). Driving such morphological changes are MAVS-dependent mitochondrial ROS generation, mitochondrial membrane hyperpolarization, inhibition of spare respiratory capacity, and modulation of ATP synthesis (see below; Buskiewicz et al., 2016; Lei et al., 2009).
An interesting link between MAVS signaling and glucose metabolism has recently been established (Fig. 2). The first step of glucose metabolism involves the phosphorylation of glucose into glucose-6 phosphate by hexokinase. Although glycolysis occurs in the cytosol, hexokinase 2 (HK2), the major enzyme that initiates glycolysis, requires mitochondrial localization for its glycolytic function (DeWaal et al., 2018; Roberts and Miyamoto, 2015; Wolf et al., 2016). The mitochondrial localization and activity of HK2 is dependent on the physical interactions of HK2 with both MAVS and VDAC situated on the OMM (Roberts and Miyamoto, 2015; Wolf et al., 2016; Zhang et al., 2019). Upon RLR activation, RIG-I binding to MAVS releases HK2 from mitochondria and thus inactivates glycolysis at its initial step (Zhang et al., 2019). On the other hand, up-regulation of glycolysis in turn suppresses MAVS signaling. Lactate, a key metabolite of anaerobic glycolysis, is a direct suppressor of RLR signaling, since it directly binds to the transmembrane domain of MAVS and prevents the formation of MAVS aggregates on the mitochondrial surface (Zhang et al., 2019).
Undesired MAVS signaling in diseases
While mitochondria play protective roles in innate immune signaling, abnormal activation of this system can nevertheless cause undesired inflammation and even cell death. For example, the integrity of the mitochondrial genome and RNAs must be tightly controlled. In situations where membrane damage leads to the leakage of mitochondrial nucleic acids, immune responses and/or cell death can be triggered (Fig. 2). Due to bidirectional transcription, mitochondrial RNAs form extensive double-stranded RNAs that are efficiently digested by the mitochondrial RNA helicase SUV3 and the 3′–5′ RNA exonuclease PNPT1 (Aloni and Attardi, 1971; Dhir et al., 2018; Young and Attardi, 1975). Hypomorphic mutations of PNPT1 result in robust accumulation and cytosolic leakage of mitochondrial double-stranded RNAs that trigger MDA5- and MAVS-dependent inflammation (Dhir et al., 2018). Additionally, mitochondrial double-stranded RNA processed by RNase L in p53-deficient cells might also trigger MAVS signaling and transcription of proinflammatory factors (Wiatrek et al., 2019). Likewise, leakage of mitochondrial DNA (mtDNA) activates the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS), leading to the further activation of stimulator of IFN genes (STING) and type I IFN production (Kim et al., 2019; West et al., 2015). Such mtDNA-stimulated innate immune signaling appears to facilitate neurodegeneration in mouse models with mitochondrial stress but defective mitophagy (Sliter et al., 2018). It would be important to further elucidate the role of inflammation mediated by mitochondrial-derived nucleic acid in other disease states.
Various RNA-independent mechanisms activating MAVS-mediated transcriptional up-regulation of proinflammatory cytokines have been described. One such mechanism involves the communication between oxidative stress and MAVS (Fig. 2). Oxidative stress is a common condition in infectious and inflammatory diseases, which can be sensed by MAVS to trigger RNA- and RLR-independent MAVS oligomerization and type I IFN production (Buskiewicz et al., 2016). Moreover, antioxidants can suppress ROS-induced MAVS oligomerization and IFN production (Buskiewicz et al., 2016), which may potentially benefit patients with autoimmune or inflammatory diseases. The fact that MAVS aggregates are resistant to detergent but sensitive to reducing agents is consistent with MAVS being an ROS sensor (Cai et al., 2014; Hou et al., 2011). In agreement with a possible role for ROS-driven disulfide bond formation in MAVS oligomerization, the MAVS-C79F variant does not oligomerize when exposed to ROS (Buskiewicz et al., 2016). Indeed, systemic lupus erythematosus patients carrying the MAVS-C79F variant show reduced plasma levels of MAVS oligomers, reduced type I IFN production, and milder disease symptoms (Buskiewicz et al., 2016; Pothlichet et al., 2011). Of note, while MAVS senses both cellular and mitochondrial ROS, MAVS aggregation can in turn promotes the generation of mitochondrial ROS and appears to be essential for infection-induced mitochondrial hyperpolarization, decrease in ATP synthesis, and drop in the spare respiratory capacity (Buskiewicz et al., 2016; Lei et al., 2009). It is still elusive how the prion-like aggregation of MAVS and the feed-forward cycle between ROS and MAVS aggregation are ultimately terminated and how mitochondrial function is restored in healthy patients after the clearance of an infection. Further mechanistic investigation of these critical deactivation steps could yield novel therapeutic strategies for autoimmune and inflammatory diseases.
Another unconventional mechanism that activates mitochondrial MAVS involves abnormal sphingolipid metabolism (Fig. 2). Sphingolipids are well known for their roles in promoting inflammation in both physiology and diseases. Expression changes of sphingolipid metabolizing enzymes have been documented upon induction of neuronal inflammation (Morita et al., 2020). In experimental autoimmune encephalomyelitis, a murine model of multiple sclerosis, the sphingolipid lactosylceramide promotes inflammation and subsequent neurodegeneration (Mayo et al., 2014). Recently, lactosylceramide has been found to trigger an interaction between MAVS and cytosolic phospholipase A2 (cPLA2) that activates MAVS aggregation and type I IFN production in astrocytes independently of RNA ligands and RLRs (Chao et al., 2019). Lactosylceramide directly binds to and activates cPLA2, a calcium-dependent phospholipase that cleaves phospholipids, releasing inflammatory lipid messengers (Chao et al., 2019; Leslie, 2015; Nakamura et al., 2013). Once activated, cPLA2 is recruited to mitochondria, where it binds and activates MAVS and changes mitochondrial metabolism by multiple mechanisms (Chao et al., 2019). First, cPLA2 binding directly triggers MAVS aggregation, leading to transcriptional production of pro-inflammatory cytokines and type I IFN. Second, cPLA2 displaces HK2 from MAVS, which causes reduced HK2 activity in glycolysis (see above) and a subsequent decline in lactate production. Third, cPLA2 activation by lactosylceramide increases the expression of cPLA2 itself and the generation of ROS, both of which, in turn, further fuel MAVS aggregation (Chao et al., 2019). Given the reported associations of MAVS signaling, cPLA2, and sphingolipids with various neurodegeneration diseases, interventions targeting the cPLA2–MAVS signaling axis might prove beneficial.
Mitochondrial retrograde signaling
Besides lipid signaling within mitochondrial membrane and innate immune signaling on the mitochondrial surface and in the cytosol, mitochondria also send out stress signals via interorganelle communication, adding another layer of potential regulation. Crosstalk between the mitochondria and nucleus is well defined in the regulation of cellular bioenergetics and stress responses. This includes anterograde signaling from the nucleus to the mitochondria, as most mitochondrial proteins are encoded by the nuclear genome, translated in the cytosol, and imported into mitochondria. On the other hand, metabolic, proteostatic or oxidative stress within mitochondria can be communicated in a retrograde fashion back to the nucleus (Fig. 3).
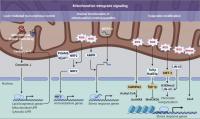
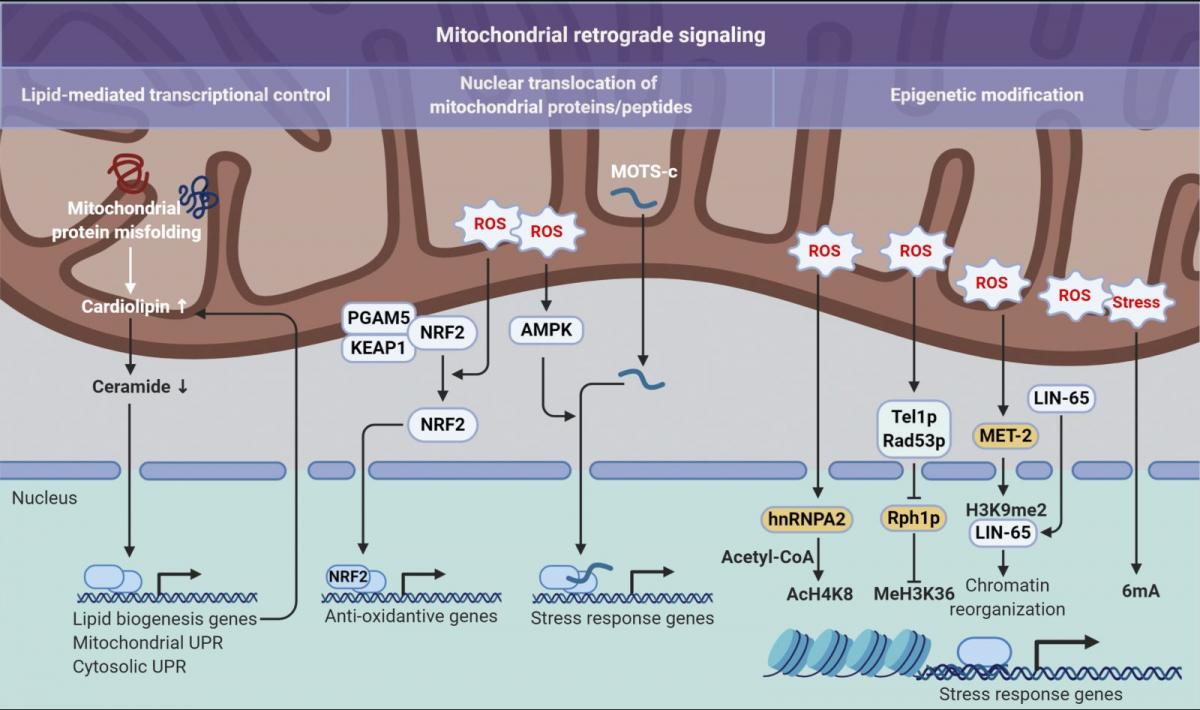
Figure 3. Mitochondrial retrograde signaling pathways. Three distinct modes of retrograde signaling that transform mitochondrial stress into nuclear programs are diagramed. On the left, mitochondrial protein folding stress is decoded into nuclear transcription via a lipid signaling-mediated UPR. In the center, mitochondrial stress provokes nuclear translocation of mitochondrial-tethered proteins or peptides that, in turn, initiate transcription of stress response genes. On the right, mitochondrial stresses, such as an increase in ROS levels, are translated into epigenetic modifications that facilitate transcriptional up-regulation of stress response genes. 6mA, N6-methyldeoxyadenine; AcH4K8, acetylation on histone H4 at lysine 8; H3K9me2, dimethylation at the ninth lysine residue of the histone H3 protein; MeH3K36, methylation on histone H3 at lysine 36.
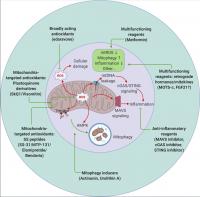
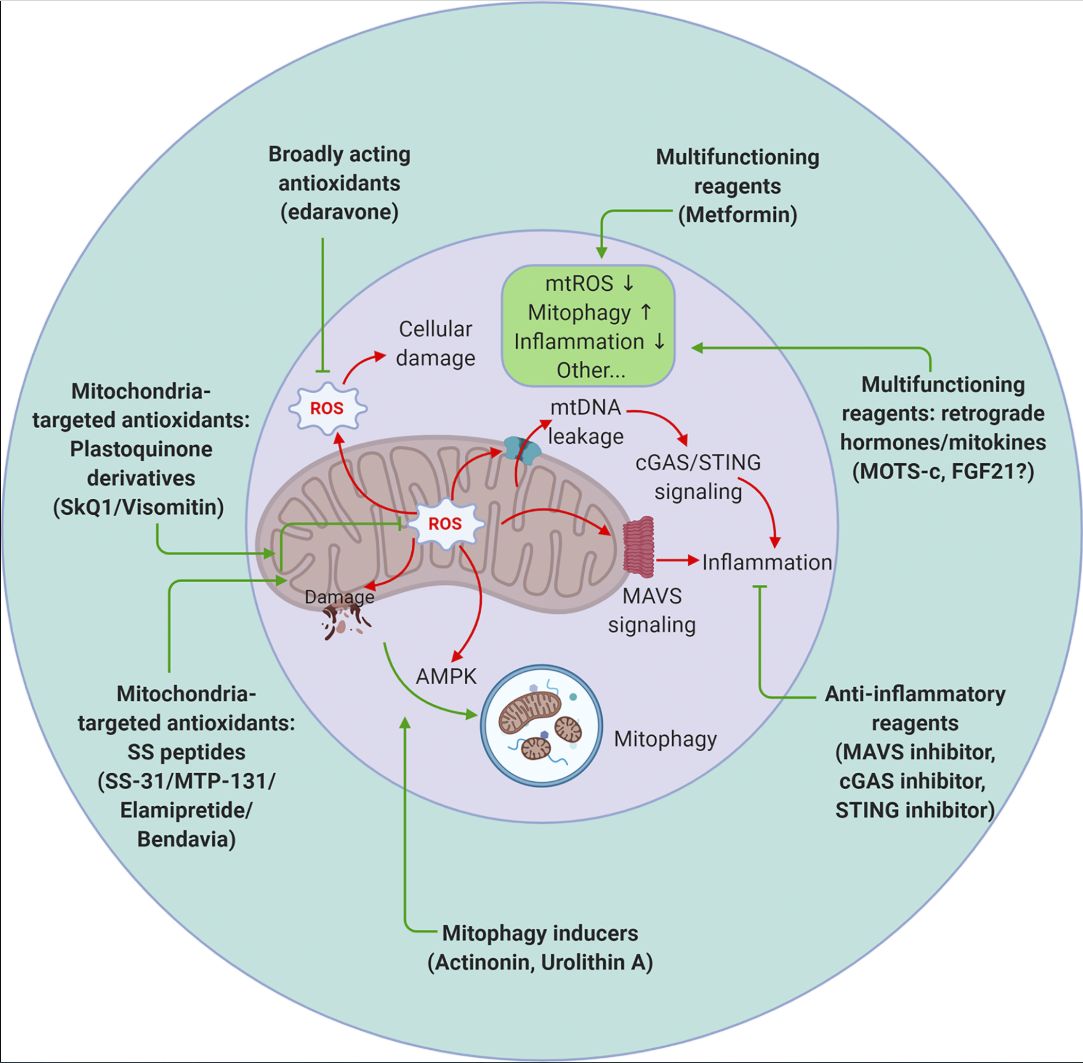
Figure 4. Therapeutic targeting of mitochondrial signaling. Strategies to target mitochondria include blocking mitochondrial damage by reducing mitochondrial ROS (antioxidants that are either broadly acting or mitochondrial specific), accelerating removal of damaged mitochondria by stimulating mitophagy (actinonin and urolithin A), and suppressing undesired inflammation caused by mitochondrial damage (inhibitors targeting the MAVS or cGAS–STING pathways). Metformin and the retrograde hormone MOTS-c appear to act through multiple mechanisms. mtROS, mitochondrial ROS.
.../...
.
Edited by Engadin, 01 May 2020 - 08:02 PM.