






























http://sens.org/
 LongeCity
LongeCityAdvocacy & Research for Unlimited Lifespans

Posted 16 July 2010 - 04:40 PM
Last summer, California-based LifeStar Institute assembled a panel of leaders in the science of aging to ask them the question at the core of their research. How far can the potential of new biomedical therapies to slow, arrest, or even reverse the damage of aging be brought to bear against the challenge of global graying?
I was honored to be brought into the midst of this remarkable assembly of scientists, who include some of the biggest names in biogerontology: names like Dr. Judith Campisi, Dr. Caleb "Tuck" Finch, SENS Foundation Chief Scientific Officer Dr. Aubrey de Grey, Dr. George Martin, and the legendary (and now, sadly, stolen from the living) Dr. Robert Butler. This is a group with a wide range of expertise and opinions within the biogerontology community, who have had strong disagreements in the past and who engaged in vigorous and stimulating debate during the meeting. But as our discussions matured, a remarkable consensus began to emerge, with the meeting ending in excitement and a sense of quiet triumph. When these experts weighed the promise of the scientific advances of recent years against the global challenge that lies before us, they came to a series of remarkable conclusions. And the most important of those conclusions was this: that an aggressive program of investment to realize that potential is not only justified, but necessary.
The conclusions of this meeting are laid out in broad detail in the ensuing meeting report: “The Demographic and Biomedical Case for Late-Life Interventions in Aging,” which has just been published in the journal Science: Translational Medicine. The headline: Aggressive biomedical research investments in new medicines to slow, arrest, and reverse the degenerative aging process are needed to turn a looming worldwide social calamity into an opportunity for a global renaissance of healthy longevity.
There is a purely moral and humanitarian case to be made for this, to be sure: the suffering and death attributable to the degenerative aging process causes enormous human suffering, in age-related disease, disability, dependence, dementia, and death. But now there is a new global social imperative as well. We stand at a cusp of an unique demographic transition. For the first time in human history, the whole planet is aging: within a few decades, people that have been made sick, dependent, or unproductive by the damage of aging will outnumber the young and healthy. The diseases of aging will rob the world of some of our most productive citizens, and rapidly drive up the cost of healthcare and the budgets for public and private pensions. Given “aging as usual,” the sheer size of the aging generation sets the stage for global economic catastrophe.
In the face of this crisis, the experts concluded, the full translation of what is known in the laboratory or foreseeable from existing biomedical developments about the damage of aging and what can be done about it into the first real "anti-aging medicines" is our best hope.
The report highlights three key approaches to the challenge that must all be met to meet the goal of maintaining the health and productivity of today’s generations: (1) expand public health measures to help citizens avoid suffering prematurely from age-related disease; (2) develop new medicines that boost the body’s ability to maintain health and productivity longer by slowing down the degenerative aging process; and (3) use the principles of regenerative engineering, the special focus of SENS Foundation, to create therapies that remove, replace, repair, and neutralize the cellular and molecular damage that accumulates in aging bodies, and thus restore youthful structure and function to the tissues and lives of aging citizens.
And to meet that goal, the report is a call for dramatic, targeted investments by the National Institutes on Health (NIH) and other public and private biomedical research organizations to bring forward new therapies against the degenerative aging process.
The time has come. The science is ready, and the need is already growing, with every aging American boomer and with the equivalent generations coming behind all over the world: in China, and in India, in Africa, in South America, and in the Middle East. The report is a call for dramatic, targeted investments by the National Institutes on Health (NIH) and other public and private biomedical research organizations to bring forward new therapies against the degenerative aging process.
"In the case of late-life intervention in human age-related degeneration, what we can be certain of today is that a policy of “aging as usual” will lead to enormous humanitarian, social, and financial costs. Efforts to avert that scenario are unequivocally merited, even if those efforts are costly and their success and full consequences uncertain. To realize any chance of success, the drive to tackle biological aging head-on must begin now."(1)
The Report
1. The Demographic and Biomedical Case for Late-Life Interventions in Aging.
Michael J. Rae, Robert N. Butler, Judith Campisi, Aubrey D. N. J. de Grey, Caleb E. Finch, Michael Gough, George M. Martin, Jan Vijg, Kevin M. Perrott, and Barbara J. Logan
Science Translational Medicine. 14 July 2010: 40cm21.
Posted 10 July 2010 - 01:03 AM
Tissue engineering and cell therapy are an essential plank in the Strategies for Engineered Negligible Senescence (SENS) platform of regenerative engineering. These biotechnologies are most obviously central for direct clinical use in repairing and replacing cells and tissues "injured by trauma, damaged by disease or worn by time" (as William Haseltine first defined regenerative medicine (1)). But additionally, mature tissue and organ engineering are a prerequisite for the building of impenetrable defenses against malignant disease in the negligibly-aging body, via the Whole-body Interdiction of Lengthening of Telomeres (WILT, or OncoSENS) strategy.(2,3) The main and earliest targets for the use of tissue engineering in WILT will be in major epithelial tissues such as the gut, skin, and lung; progress toward tissue engineering of these organs is therefore of higher priority for SENS Foundation than would be expected from direct projections from existing clinical needs for transplant medicine.
A recent report from researchers at Yale and Duke Universities (4) heralds a significant advance toward the tissue engineering of the lung.
.... we treated lungs from adult rats using a procedure that removes cellular components but leaves behind a scaffold of extracellular matrix that retains the hierarchical branching structures of airways and vasculature. [Compare the pioneering work of Dr. Doris Taylor's group in the rat heart,(5) and of Macchiarini et al in the bronchus (6) -MR] We then used a bioreactor to culture [neonatal] pulmonary epithelium and vascular endothelium on the acellular lung matrix. The seeded epithelium displayed remarkable hierarchical organization within the matrix, and the seeded endothelial cells efficiently repopulated the vascular compartment. In vitro, the mechanical characteristics of the engineered lungs were similar to those of native lung tissue ...
[W]hen implanted into rats in vivo for short time intervals (45 to 120 min), the engineered lungs participated in gas exchange. ... In all cases, the engineered lungs were easily suturable to the recipient and they were ventilated with no visible air leak from the parenchyma. All engineered lungs became perfused with blood over a period of seconds to minutes, with blood visibly turning from dark to bright red as the hemoglobin became oxygenated. ... After perfusion and ventilation, blood gas samples were drawn from the pulmonary artery, left and right pulmonary veins [individually], ... and from the unclamped pulmonary vein, to document the extent of gas exchange ... [T]he engineered lung was inflated with air, but the level of inflation was less than that of the native right lung. ... Partial pressures of oxygen increased from 27±7 mmHg in the pulmonary artery, to 283±48 mmHg in the left pulmonary vein ... Although the partial pressure of oxygen in the right pulmonary vein was higher [634±69 mmHg], ... this difference may not be of substantial physiological consequence, since hemoglobin saturation is complete above oxygen pressures of 100 mm [and was 100% for both venous samples in this study] ... In addition, carbon dioxide removal was efficient, with CO2 falling from 41±13 mmHg in the pulmonary artery to 11±5mmHg in the left, engineered pulmonary vein. [As with p02, pCO2 of the native right lung's pulmonary vein was approximately half of that in the left, engineered one; the mixed venous blood was roughly halfway between that of the right and left pulmonary veins considered in isolation. As a nonspecialist, I do not feel qualified to speculate on the functional significance of these nominal deficiencies beyond the authors' cautious reasurrances -MR]. ...
Although representing only an initial step toward the ultimate goal of generating fully functional lungs in vitro, these results suggest that repopulation of lung matrix is a viable strategy for lung regeneration.
This represents a substantial advance for tissue engineering via the repopulated stromal scaffold approach, executed in a tissue far more complex in structure and function than the bronchus (6) and actually shown to function, albeit only for a brief window, in vivo rather than demonstrated ex vivo (5). And as noted, the engineering of functional lungs with autologous cells is of particular importance to WILT (2,3). The broad outlines of the path ahead for clinical use to replace whole lung transplantation in injury and pulmonary disorders is reasonably clear, moving through ongoing refinement of the protocol and its demonstration for progressively longer periods of time, to translating the technique first to large mammal models, and later to human patients. The latter might initially be achieved via the use of decellularized porcine or other lung tissue as a xenoscaffold with autologous patient cells. Later, further experience and innovation in tissue engineering, along with greater understanding of the stroma (including its development, the "body as best bioreactor," and its interactions with lung parenchyma) should allow for completely engineered scaffolds to take the place of biologically-sourced ones. The eventual engineering of cancer-impervious lung tissue will require, in addition, the generation of suitable patient-derived engineered cells, with the telomere maintenance machinery deleted and telomeres lengthened to youthful physiological levels ex vivo, and , and their use in the seeding of such scaffolds. If scaffold technology is sufficiently sophisticated at that time, the engineering of aditional cell populations responsible for the ongoing physiological maintenance of the engineered stroma in situ may also be desirable. This landmark report is a step change along that path.
References
1. Haseltine WA. The emergence of regenerative medicine: a new field and a new society. J Regen Med. 2001 Jun 7;2(4):17.
2. de Grey ADNJ, Campbell FC, Dokal I, Fairbairn LJ, Graham GJ, Jahoda CAB, Porter ACG. Total deletion of in vivo telomere elongation capacity: an ambitious but possibly ultimate cure for all age-related human cancers Ann N Y Acad Sci. 2004 Jun;1019:147-70. PubMed: 15247008.
3. de Grey ADNJ. Whole-body interdiction of lengthening of telomeres: a proposal for cancer prevention. Front Biosci 2005;10:2420-2429. PubMed: 15970505.
4. Petersen TH, Calle EA, Zhao L, Lee EJ, Gui L, Raredon MB, Gavrilov K, Yi T, Zhuang ZW, Breuer C, Herzog E, Niklason LE. Tissue-Engineered Lungs for in Vivo Implantation. Science. 2010 Jun 28. [Epub ahead of print] PubMed PMID: 20576850.
5. Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, Taylor DA. Perfusion-decellularized matrix: using nature's platform to engineer a bioartificial heart. Nat Med. 2008 Feb;14(2):213-21. Epub 2008 Jan 13. PubMed PMID: 18193059.
6. Macchiarini P, Jungebluth P, Go T, Asnaghi MA, Rees LE, Cogan TA, Dodson A, Martorell J, Bellini S, Parnigotto PP, Dickinson SC, Hollander AP, Mantero S, Conconi MT, Birchall MA. Clinical transplantation of a tissue-engineered airway. Lancet. 2008 Dec 13;372(9655):2023-30. Epub 2008 Nov 18. Erratum in: Lancet. 2009 Feb 7;373(9662):462. PubMed PMID: 19022496.
Posted 16 June 2010 - 12:29 AM
As anyone following the field will know, the derivation of induced pluripotent (iPS) cells reprogrammed from differentiated somatic cells offers a remarkable promise: the ability to generate donor-specific pluripotent stem cells, without the "ethical" confusion that has so unfortunately retarded the progress of somatic cell nuclear transfer (SCNT) research. However, protocols to date have not led to systems that could yet viably be scaled up into therapeutic use: efficiency has been extremely low, and the reliance on the proto-oncogene c-Myc and/or of viral vectors in such systems have made the risk of transformation too high to realistically be tested in humans. A more detailed understanding of the mechanistic basis for reprogramming could potentially allow for the bypassing of these steps, and/or increases in efficiency, that would bring therapies for the degenerative processes of aging and a range of injuries and diseases closer to the clinic.
We were therefore surprised to note how little attention has been given to a recent report in Nature(1) from the Blau lab at Stanford's Institute for Stem Cell Biology and Regenerative Medicine. In the few commentaries that have appeared online, either offhanded allusion or no discussion at all is given to the potential that these findings could lead to tools that would greatly increase the efficiency of cellular reprogramming, although a recent review(7) was more appropriately enthusiastic in reviewing this and a related report (9).
Their group and others had previously shown that fusing mouse ESC with human fibroblasts led to heterokaryons led to induction of previously-silent genes that allowed the cells, like iPS cells, to take on other differentiated cell fates. They "showed that reprogramming in heterokaryons was influenced by DNA methylation status, tissue of origin, and the relative ratio of nuclei that dictates the balance of regulators, consistent with recent experiments in iPS cells." (1) In the present study, they exploited this system
to study epigenetic and transcriptional changes critical to the initiation of reprogramming towards pluripotency. We focused on DNA demethylation—a known block to reprogramming that leads to partially reprogrammed iPS cells, and also a key step for reprogramming by nuclear transfer. Despite decades of effort, so far no consensus mammalian DNA demethylase has been identified. Recently, [Activation-Induced (Cytidine) Deaminase (AID)] has been implicated in DNA demethylation in zebrafish within hours after fertilization, acting in a complex that mediates deamination followed by DNA repair. In mammals, AID is primarily known for its role in the generation of antibody diversity in B lymphocytes, but has recently been detected in germ cells. (1)
Indeed, in addition to germ cells,(2,3) AID had been found to be activated in oogenesis and early development,(4) including during spermatogenesis in both normal and telomerase-disabled (TERC-/-) mice.(5)† It was therefore a reasonable candidate for involvement in maintenance or induction of pluripotency. The hetereokaryons seemed likely to be a good system in which to study such changes during the presumed reprogramming process because interspecies differences would allow them to distinguish between expression changes in gene transcripts derived from the two parent cell types; moreover, the process does not involve cell division, allowing them to rule out the possibility that any observed CpG demethylation might be the result of stochastic errors in methylation maintenance after replication of DNA during S phase.
(siRNA)... showed that... AID ... is required for promoter demethylation and induction of OCT4 (also known as POU5F1) and NANOG gene expression. AID protein bound silent methylated OCT4 and NANOG promoters in fibroblasts [and the homeobox protein Cdx2 in mouse ESC], but not active demethylated promoters in ES cells. These data provide new evidence that mammalian AID is required for active DNA demethylation and initiation of nuclear reprogramming towards pluripotency in human somatic cells.(1)

Figure: "Model for AID-dependent active DNA demethylation in reprogramming. ... The other putative components of this mammalian DNA demethylase complex (X, Y and Z) that may act together with the deaminase, AID, remain to be identified. 5mC, 5-methyl-cytosine." From (1).
As was noted by a commentary by Drs. Suneet Agarwal and George Q Daley,
Several important questions remain: (1) Are the same mechanisms at work during reprogramming by NT [somatic cell nuclear transfer] or in the generation of iPS cells? The possibility is favored by the observations of active demethylation of the paternal genome after natural fertilization, and the facilitation of iPS cell generation using agents that interfere with maintenance of DNA methylation. ... The results of Bhutani et al. suggest that AID function is not replaceable by other cytidine deaminase family members. Therefore, it would be interesting ... to assess the effects of AID disruption on human iPS cell generation ... (2) What are the mechanisms activating AID during reprogramming? The observation by Bhutani et al. that AID binds to the OCT4 and NANOG promoters in fibroblasts, where the loci remain methylated, implies that the presence of AID is not sufficient for demethylation. Factors either resident in mouse ES cells or induced in the heterokaryons must therefore be invoked to explain AID-mediated activation of demethylation at these loci after fusion.(8)
Again, we are surprised by the mild tone and narrowly-defined discussion of the potential of this report. Perhaps the remarkable rush of research monies and investigators into the iPS field, and the ensuing breathtaking pace of progress in iPS research that began in 2008 and has continued ever since, has led to some unfortunate fatigue with the sheer volume of reports. Indeed, the very same issue of Nature also featured a supporting report that "Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency,"(9) and the potentially-useful finding that "Tbx3 improves the germ-line competency of induced pluripotent stem cells"(10) (which seems to have garnered more enthusiasm for its clinical potential). Whatever the reason for the blasé response, mall molecules or other convenient and readily-reversible inducers of the same processes could reasonably be anticipated to lead to a similar leap in efficiency and rapidity of reprogramming similar to that observed in the Blau group's heterokaryons, bringing the safe and scalable production of patient-specific pluripotent cells needed for cell therapy in tissue engineering for biomedical rejuvenation closer to reality.
†The expression of AID during spermatogenesis in telomerase-deficient mice,(5) combined with its involvement in somatic hypermutation and immunoglobulin class switching in B-cells, and the observed elongation of B-cell telomeres in a heterogeneous pattern in the same model following immunization(6) and several other findings, led Dr. de Grey to propose that aberrant AID expression might be a key component of the ALT telomere-lengthening mechanism. SENS Foundation funded preliminary studies to probe this possibility by Dr. F. Mathias Bollmann at Universitätsklinikum Hamburg-Eppendorf; these studies appear to have discounted AID's involvement (unpublished results; personal communication, M. Bollmann).
References
1: Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010 Feb 25;463(7284):1042-7. PubMed PMID: 20027182.
2: Schreck S, Buettner M, Kremmer E, Bogdan M, Herbst H, Niedobitek G. Activation-induced cytidine deaminase (AID) is expressed in normal spermatogenesis but only infrequently in testicular germ cell tumours. J Pathol. 2006 Sep;210(1):26-31. PubMed PMID: 16783758.
3. Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004 Dec 10;279(50):52353-60. Epub 2004 Sep 24. PubMed PMID: 15448152.
4. Liu L, Bailey SM, Okuka M, Muñoz P, Li C, Zhou L, Wu C, Czerwiec E, Sandler L, Seyfang A, Blasco MA, Keefe DL. Telomere lengthening early in development. Nat Cell Biol. 2007 Dec;9(12):1436-41. Epub 2007 Nov 4. PubMed PMID: 17982445.
5. Tanemura K, Ogura A, Cheong C, Gotoh H, Matsumoto K, Sato E, Hayashi Y, Lee HW, Kondo T. Dynamic rearrangement of telomeres during spermatogenesis in mice. Dev Biol. 2005 May 15;281(2):196-207. PubMed PMID: 15893973.
6. Herrera E, Martínez-A C, Blasco MA. Impaired germinal center reaction in mice with short telomeres. EMBO J. 2000 Feb 1;19(3):472-81. PubMed PMID: 10654945; PubMed Central PMCID: PMC305584.
7. Deng W. AID in reprogramming: quick and efficient: identification of a key enzyme called AID, and its activity in DNA demethylation, may help to overcome a pivotal epigenetic barrier in reprogramming somatic cells toward pluripotency. Bioessays. 2010 May;32(5):385-7. PubMed PMID: 20394066.
8. Agarwal S, Daley GQ. AID for reprogramming. Cell Res. 2010 Mar;20(3):253-5. PubMed PMID: 20190775.
9. Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010 Feb 25;463(7284):1101-5. PubMed PMID: 20098412.
10. Han J, Yuan P, Yang H, Zhang J, Soh BS, Li P, Lim SL, Cao S, Tay J, Orlov YL, Lufkin T, Ng HH, Tam WL, Lim B. Tbx3 improves the germ-line competency of induced pluripotent stem cells. Nature. 2010 Feb 25;463(7284):1096-100. Epub 2010 Feb 7. PubMed PMID: 20139965.
Posted 08 June 2010 - 01:11 AM
In late 2008, we reviewed then-unpublished work by Dr. Mark Pepys, who was working on an ambitious project anticipated to allow for the disaggregation of nearly all disease-associated amyloids. Dr. Pepys subsequently accepted an invitation to present those early results at the fourth SENS scientific conference(1). His strategy is based on the fact that the pentraxin serum amyloid P component (SAP) is an "universal constituent of the abnormal tissue deposits in amyloidosis, including Alzheimer disease". (2) As we reviewed:
A quarter century ago, Pepys suggested that because circulating SAP is believed to exist in a state of dynamic equilibrium with the SAP in amyloid deposits, lowering circulating SAP might lead plaque SAP to dissociate, leading to the breakup of the integrity of the plaque and ultimate clearance of amyloid deposits.
Early in this century, Pepys' team began ... search[ing for] a small molecule that might inhibit the binding of SAP in Abeta, and came across one that was particularly effective: R-1-[6-[R-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl] pyrrolidine-2-carboxylic acid, or CPHPC, which "also crosslinks and dimerizes SAP molecules",([3]) blocking the binding face of the molecule in the process. They quickly moved from this in vitro finding through animal studies for efficacy and toxicity, and the initial results being favorable, moved into pilot human studies.([4])
In 2002, Pepys reported that adminstration of IV CPHPC over the course of 2 days into 8 amyloidosis patients resulted in almost total removal of SAP from the circulation, apparently through "very rapid" hepatic clearance, since tracer studies found a large amount of SAP in the liver of one patients 6 h after initiating treatment.([4]) They quickly sent CPHC sailing down the drug-development pipeline, to results that, although still preliminary, were extremely exciting.
The promise
We reviewed some preliminary findings from subsequent pilot studies in light chain (AL) amyloidosis and other diseases, some of which were later reported and elaborated at SENS4 -- including very brief allusion to some work on AD patients. To surprisingly little fanfare (so little that it was not noticed by the present author), Dr. Pepys reported results from those pilot studies late in 2009:
We therefore conducted a pilot proof of concept study in 5 patients aged 53–67 years with mild to moderate probable Alzheimer's disease who received 60 mg CPHPC by s.c. injection 3 times daily for 12 weeks. The drug was well tolerated with no adverse effects other than transient local discomfort on injection. Compliance was confirmed by the presence of CPHPC in all serum samples ... [and] in the cerebrospinal fluid. The concentration of SAP in the serum fell dramatically ... 1 week after starting CPHPC, remained around this value throughout the treatment period, and had [normalized] ... at 4 weeks after drug discontinuation (Fig. 1B). The SAP concentration in the CSF also fell remarkably [my emphasis], ...and then remained scarcely detectable throughout the treatment period. Because depletion of circulating SAP by CPHPC involves clearance by the liver, these observations ... suggest that entry of CPHPC into the brain is not required for removal of intracerebral SAP but the presence of CPHPC in CSF should additionally block effects of any residual or locally produced SAP.
There was no significant difference between the clinical measures before and after CPHPC and importantly no deterioration in [cognitive scores] ... nor any structural change in MRI brain scans... [nor] in CSF concentrations of Aβ40, Aβ42, total or phosphorylated tau, or S100B or in any of the comprehensive routine hematological, biochemical, endocrine, or serological blood tests. ... [I]t would have been astonishing if there had been any improvement in cognition or change in the CSF biomarkers during this brief study ... However, the clinical stability and absence of biochemical signs of cerebral damage importantly confirm the safety in patients with dementia both of CPHPC itself and of profound depletion of systemic and cerebral SAP and support longer-term studies of clinical efficacy.(5)
As Pepys also notes, there are additional possible pathogenic roles for SAP in AD beyond their direct effect in Aß aggregation:
the universal presence of serum amyloid P component (SAP) in cerebrospinal fluid (CSF) and bound to cerebral and cerebrovascular amyloid deposits and to neurofibrillary tangles in Alzheimer disease [my emphasis] is consistent with a role of SAP in pathogenesis. .... Furthermore, human SAP has been reported to bind to and enter neurons in culture and in rat brain in vivo, to cause apoptotic cell death, and to activate human microglia synergistically with Aβ and C1q [required for immune complex binding to phagocytes] in vitro, provoking increased production of pro-inflammatory cytokines and Aβ itself. The presence of tangles composed of hyperphosphorylated tau protein, to which SAP binds in non-Alzheimer dementias, also raises the possibility of targeting SAP in those conditions.(5)
While preliminary, these results are promising, and we are pleased to see the work published and advancing. However, there is also reason for caution. At its core, the therapeutic use of CPHPC is based on what the authors rightly characterize as its ability to induce the "unprecedented, profound depletion" (by >99%) of a physiological protein from the systemic circulation and CSF. Few side-effects have been observed in the small human studies conducted to date,(4,5) but these studies have been performed in patients rendered very ill by massive, prevalent accumulations of a single -- and, in most patients' case, a mutant -- protein. If SAP depletion is to be used to target the more gradual, ongoing accumulation of multiple distinct species of extracellular aggregates formed from wild-type proteins during aging, its physiological roles will ipso facto be arrested for an extended percentage of the year, for each year of a current adult life expectancy and beyond. As with all such "gerontological" interventions against aging, such interference in body's homeostasis can be expected to come with significant potential for adverse outcomes -- and it is exceptionally difficult to predict the unintended side-effects of extended intermittent interference in the case of SAP, as its physiological functions (such as its complex dual role in bacterial immunity(6)) are as yet little-understood.
As it happens, an hypothetical complication of chronic SAP suppression, conducted even intermittently over many decades, has just emerged.
The peril?
A group of researchers at the Harvard Institutes of Medicine and Harvard Medical School recently became interested in the possible role of SAP in controlling the fibrotic response to tissue injury.
[The] unique binding activities of SAP and in vitro biology studies suggest that SAP may localize specifically to sites of injury and aid in the removal of damaged tissue and pathogenic organisms. ... Despite extensive characterization of SAP in vitro, its potential participation in natural regulation of the innate injury response has only recently been appreciated. ... Because FcγR [Fcγ receptor] expression is restricted predominantly to cells of the innate immune system, and many of the ligands for SAP are concentrated at sites of tissue injury, we predicted that SAP binding to ligands might affect innate immune cell activation events in a localized fashion and thereby potentially modulate the innate injury response. ...
Pilling et al. ... showed that purified rat SAP could suppress development of lung fibrosis in the bleomycin model, which correlated with reduced fibrocyte numbers within the lung tissue. However, fibrocytes play no obvious role in the development of fibrosis of the kidney; therefore, we wished to determine whether SAP would have an antifibrotic effect in this tissue setting and, if so, what mechanisms mediated its biologic effect....
Here we show that fibrosis progression in the mouse kidney is significantly inhibited by therapeutic administration of human serum amyloid P, regulated by activating Fcγ receptors, and dependent on inflammatory monocytes and macrophages, but not fibrocytes. Human serum amyloid P-mediated inhibition of mouse kidney fibrosis correlated with specific binding of human serum amyloid P to cell debris and with subsequent suppression of inflammatory monocytes and kidney macrophages in vitro and in vivo, and was dependent on regulated binding to activating Fcγ receptors and interleukin-10 expression.
These studies uncover previously unidentified roles for Fcγ receptors in sterile inflammation and highlight serum amyloid P as a potential antifibrotic therapy through local generation of interleukin-10.[emphasis mine](7)
This is an important finding in its own right, as SAP's physiological function is currently so poorly-understood. But from the viewpoint of biomedical gerontology, an antifibrotic role of SAP suggests a possible long-term risk associated with suppression of this key immunological protein as a potential prophylactic against the damage of aging. In addition to Aß, an universal anti-amyloid therapeutic would potentially be used to target a wide swath of age-related extracellular aggregates, including notably the age-related cardiac amyloidoses, which have emerged as critical-path targets for regenerative engineering.
The prevalence of age-related cardiac amyloidoses (notably senile cardiac amyloidosis (caused by aggregated wild-type transthyretin (TTR)) and isolated atrial amyloidosis (IAA) (caused by aggregated atrial natriureptide (ANP)) rises dramatically late in a currently-normal life expectancy(9,10) and appears to be responsible for, or a significant contributor to, a large number of deaths in centenarians and especially supercentenarians.(11) At the same time, fibrosis and "ectopic" collagen infiltration are prominent findings in the aging myocardium.(8) Cardiac remodeling replaces lost cardiomyocytes with fibrotic tissue in order to preserve the gross structural integrity of the aging heart, but as (7) notes absent this context, "Fibrosis itself causes parenchymal cell ischemia, distortion, and contraction of normal organ architecture and contributes directly to functional demise." A parallel statement could readily be inserted in the opening paragraphs of a report on age-related cardiac amyloidosis. Intermittent inhibition of the organism's ability to counterregulate the fibrotic response to injury in the heart could reasonably be hypothesized to accelerate this age-related degeneration of tissue architecture. And clearly, the brief months of clinical trials of SAP clearance with CPHPC to date(3-5) would not be sufficient to reveal any such long-term acceleration of this pathological structural decay, particularly in biologically aged patients with severe pre-existing pathology,
Of note, upon being informed of this new report,(7) Stan Primmer of the Supercentenarian Research Foundation (SRF) disclosed to the present author (personal communication, 2010-05-31 9:59 PM) that the autopsies of several supercentenarians have revealed the occurrence of fibrosis; in fact, one recent supercentenarian death could be attributed to pneumonia due to idiopathic pulmonary fibrosis -- and, by coincidence or not, this subject had an uncharacteristic lack of amyloidosis. While a single case report cannot be counted as evidence of any merit, this finding would be consistent with the idea that low levels or activity of endogenous SAP would at once result in slower progression of age-related amyloidoses, while simultaneously accelerating the deposition of fibrotic tissue by limiting the body's ability to counterregulate the fibrotic response in pulmonary and other tissue. Irrespectively, the reasonable concern that repeated bouts of active pharmacological suppression of SAP for many decades would haunt its clinical use to retard or arrest the accumulation of age-related amyloidoses.
A Planned Collaboration in Rejuvenation Engineering
The "engineering" heuristic of anti-aging biomedicine is to target not the metabolic basis of age-related pathology, but the inert damage of aging itself. I am therefore delighted to have the privilege to be given permission by Stan Primmer to make the first public announcement that the SRF has recently helped to facilitate a collaboration between Drs. Sudhir Paul and Brian O’Nuallain, researchers already working in amyloid diseases, to develop antibodies to cleave aggregated wild-type and mutant transthyretin -- the form responsible for senile cardiac amyloidosis (a prevalent, but not exclusive, cardiac amyloidosis in supercentenarians).
The project will proceed in four phases. Phase 1 will consist of in vitro generation of ... monoclonal... catalytic antibodies that can directly destroy TTR amyloid ... After the initial breakdown cycle, the catalytic antibody would be re-used again and again to disintegrate additional TTR molecules. Thus, only a small amount of the catalytic antibody should be needed to remove large amounts of TTR amyloidogenic precursors and TTR amyloid. ... Phase 2 will consist of studies in an animal model of TTR amyloidosis to determine the safety and efficacy of the potential ... therapeutic antibodies discovered in Phase 1. Phase 3 will involve clinical trials on the diagnostic and therapeutic potential of the novel antibodies for comparatively young human subjects who develop amyloidosis due to mutation(s) in the TTR molecule. Initial testing of the methodology in the younger cohort is designed to avoid risk to the uniquely fragile group consisting of supercentenarians. Phase 4 will then serve to apply the results of previous research to volunteer supercentenarians in order to improve their health and extend their lives beyond what they would otherwise be expected to live.(11)
SENS Foundation recognizes the importance of this project. In conception and ambition, this collaboration offers the potential to be exceptionally exciting, pioneering research, probing the extremes of human longevity with a bold new therapeutic strategy. Actual implementation of this proposal currently awaits the availability of funding, which has not yet been secured. At this time, we can only wish these experienced specialists good luck and godspeed.
References
1.Pepys M. Treatment and prevention of amyloidosis. Rejuvenation Res. 2009;12 (Suppl 1):S46–S47.
2. Pepys MB, Rademacher TW, Amatayakul-Chantler S, Williams P, Noble GE, Hutchinson WL, Hawkins PN, Nelson SR, Gallimore JR, Herbert J, et al. Human serum amyloid P component is an invariant constituent of amyloid deposits and has a uniquely homogeneous glycostructure. Proc Natl Acad Sci U S A. 1994 Jun 7;91(12):5602-6. PMID: 8202534 [PubMed - indexed for MEDLINE]
3. Pepys MB. Science and serendipity. Clin Med. 2007 Dec;7(6):562-78.Links PMID: 18193704
4. Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, Lovat LB, Bartfai T, Alanine A, Hertel C, Hoffmann T, Jakob-Roetne R, Norcross RD, Kemp JA, Yamamura K, Suzuki M, Taylor GW, Murray S, Thompson D, Purvis A, Kolstoe S, Wood SP, Hawkins PN. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002 May 16;417(6886):254-9. PMID: 12015594 [PubMed - indexed for MEDLINE]
5. Kolstoe SE, Ridha BH, Bellotti V, Wang N, Robinson CV, Crutch SJ, Keir G, Kukkastenvehmas R, Gallimore JR, Hutchinson WL, Hawkins PN, Wood SP, Rossor MN, Pepys MB. Molecular dissection of Alzheimer's disease neuropathology by depletion of serum amyloid P component. Proc Natl Acad Sci U S A. 2009 May 5;106(18):7619-23. Epub 2009 Apr 16. PubMed PMID: 19372378; PubMed Central PMCID: PMC2669789.
6. Noursadeghi M, Bickerstaff MC, Gallimore JR, Herbert J, Cohen J, Pepys MB. Role of serum amyloid P component in bacterial infection: protection of the host or protection of the pathogen. Proc Natl Acad Sci U S A. 2000 Dec 19;97(26):14584-9. PubMed PMID: 11121061; PubMed Central PMCID: PMC18962.
7. Castaño AP, Lin SL, Surowy T, Nowlin BT, Turlapati SA, Patel T, Singh A, Li S, Lupher ML Jr, Duffield JS. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Transl Med. 2009 Nov 4;1(5):5ra13. Erratum in: Sci Transl Med. 2009 Nov 4;1(5):5ra13. PubMed PMID: 20368175; PubMed Central PMCID: PMC2852889.
8. de Souza RR. Aging of myocardial collagen. Biogerontology. 2002;3(6):325-35. Review. PubMed PMID: 12510171.
9. Kholová I, Niessen HW. Amyloid in the cardiovascular system: a review. J Clin Pathol. 2005 Feb;58(2):125-33. Review. PubMed PMID: 15677530; PubMed Central PMCID: PMC1770576.
10. Steiner I, Hájková P. Patterns of isolated atrial amyloid: a study of 100 hearts on autopsy. Cardiovasc Pathol. 2006 Sep-Oct;15(5):287-90. 16979036
11. Primmer SR, Paul S, O’Nuallain B. Research project to extend lives of supercentenarians by diagnosing and treating transthyretin amyloidosis. Unpublished MS, Supercentenarian Research Foundation. 2010 Mar 25.
Posted 28 May 2010 - 12:09 AM
In a previous update, we reviewed a recent report from a group looking to select the most active beta-amyloid (Abeta)-targeting antibodies from pooled human immunoglobulin for injection (IVIgG). As we noted there,several small, early-phase clinical trials of IVIgG for Alzheimer's disease have reported promising results. Moreover, the IVIgG preparations used in these trials are already available and approved for other indications. Baxter Pharma has announced that they will cosponsor Phase III trials their Gammagard IVIgG for Alzheimer's in conjunction with the NIH, and Octapharma is now recruiting subjects for a Phase II trial of Octagam IVIgG.
There are doubtless only a few antibodies in the mixed IVIgG pool that are responsible for the Abeta-clearing effect. It is reasonable to assume that there will be significant batch-to-batch variability in the concentration of these active antibodies, and that the other antibodies will exert at least some undesirable off-target effects. Thus, the identification of the key species responsible for Abeta removal and clinical benefits would allow for the creation of more effective therapeutics, which could be manufactured in consistent lots through recombinant DNA, improving safety and efficacy and increasing the supply of what is currently an extremely costly therapeutic, due to the inherent limits of imposed by sourcing IVIgG from pooled human plasma.
A recent report further documents the existence and possible physiological significance of these Abs, and may offer significant potential for their identification, isolation, and therapeutic exploitation.
Here, we demonstrate with peptide microarrays the presence of natural antibodies against known toxic Abeta and amyloidogenic non-Abeta species in plasma samples and cerebrospinal fluid of AD [Alzheimer disease] patients and healthy controls aged 21-89 years.
Antibody reactivity was most prominent against oligomeric assemblies of Abeta and pyroglutamate or oxidized residues [soluble Abeta species most implicated in Abeta-related synaptic dysfunction and cognitive deficits -MR], and IgGs specific for oligomeric preparations of Abeta1-42 in particular declined with age and advancing AD. Most individuals showed unexpected antibody reactivities against peptides unique to autosomal dominant forms of dementia (mutant Abeta, ABri, ADan) and IgGs isolated from plasma of AD patients or healthy controls protected primary neurons from Abeta toxicity.
Aged vervets showed similar patterns of plasma IgG antibodies against amyloid peptides, and after immunization with Abeta the monkeys developed high titers not only against Abeta peptides but also against ABri and ADan peptides. [This provides a useful and rapidly-available primate model for the evaluation of any resulting monoclonal Abeta immunotherapy -MR].
Our findings support the concept of conformation-specific, cross-reactive antibodies that may protect against amyloidogenic toxic peptides. If a therapeutic benefit of Abeta antibodies can be confirmed in AD patients, stimulating the production of such neuroprotective antibodies or passively administering them to the elderly population may provide a preventive measure toward AD. (1)
Reference
1: Britschgi M, Olin CE, Johns HT, Takeda-Uchimura Y, LeMieux MC, Rufibach K, Rajadas J, Zhang H, Tomooka B, Robinson WH, Clark CM, Fagan AM, Galasko DR, Holtzman DM, Jutel M, Kaye JA, Lemere CA, Leszek J, Li G, Peskind ER, Quinn JF, Yesavage JA, Ghiso JA, Wyss-Coray T. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer's disease. Proc Natl Acad Sci U S A. 2009 Jul 21;106(29):12145-50. Epub 2009 Jul 6. PubMed PMID: 19581601; PubMed Central PMCID: PMC2715538.
Posted 16 May 2010 - 12:29 AM
Aggregates of beta-amyloid (Aß) and other malformed proteins accumulate in brain aging and neurodegenerative disease, leading progressively to neuronal dysfunction and/or loss. The regenerative engineering solution to these insults is therapeutic clearance of aggregates, extracellular (such as Aß plaques) and intracellular (such as soluble, oligomeric Aß). Immunotherapeutic Aß clearance from the brain is a very active field of Alzheimer's research, with at least seven passive, and several second-generation active, Aß vaccines currently in human clinical trials.(1)
One challenge to optimal vaccine design is matching the specificity of antibodies tthe range of Aß aggregates that form in vivo, including different oligomeric and protofibrillar assemblies in addition to the Aß fibrils that compose the hallmark plaques of aging Alzheimer's disease (AD) brain. Research has shown that agents that sequester one Aß species may leave other species intact, and in some cases a shift in assembly dynamics can actually promote the formation of one species while clearing or reducing the formation of others (eg, (2-6)). On the other hand, insufficiently Aß-specific antibodies may target the physiological amyloid precursor protein (APP). While the balance of evidence suggests that toxic oligomeric species are the most impairing Aß assembly, and while some researchers even believe that the insoluble plaques may act as protective "sinks" that sequester more toxic Aß species, all such aggregates are stochastic accretions to the aging brain, and the ideal Aß-targeting strategy would lead to the clearance of all Aß aggregates while not intervinging with APP metabolism.
Although in very early in vivo testing, a new approach has emerged that may offer that promise. This is the use of an Aß-targeting affibody, ie, a novel non-immunoglobulin binding protein generated through combinatorial protein engineering, using phage display to select ligands from a non-cysteine three-helix bundle domain library.(7) In 2007, a team from Sweden's Royal Institute of Technology (KTH) reported the generation the selective affibody ZAβ3, which forms a disulfide-linked dimer that binds with nanomolar affinity to Aß monomers.(8) Upon binding to Aß, the ZAβ3 dimer forms stable complexes with Aß, forcing it into a hairpin conformation and safely burying its hydrophobic, aggregation-prone domains within its structure (Fig 1).
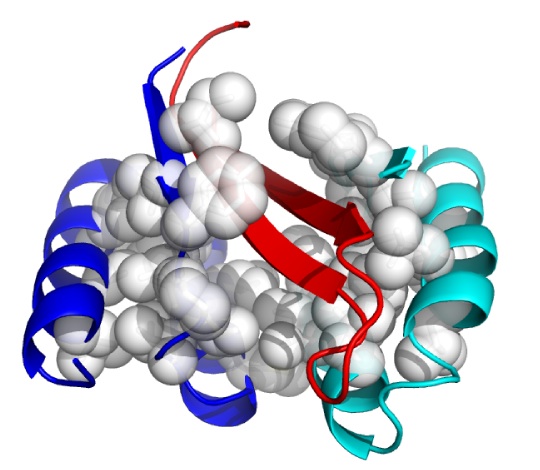
Figure 1: ZAβ3 (blue and cyan; nonpolar side chains in white) complexed to an Aβ40 hairpin at residues 16-40 (red). From (9).
To test the effects of ZAβ3 on Aß aggregatess and neurotoxicity in vivo, Swedish and English collaborators used Drosophila lines transgenically expressing either wild-type Aß42, E22G (the preferrentially oligomer-forming "Arctic" Aß mutation first identified in a Swedish familial AD proband), or the relatively benign, non-aggregative isoform Aß40. These were then crossed with strains transgenic for ZAβ3; for 2 copies of ZAβ3 connected head-to-tail ((ZAβ3)2) to facilitate formation of the ZAβ3 dimer; or for overexpression of the wild-type Z domain alone (as controls). The resulting hybrids were crossed onto strains bearing drivers to ensure coexpression of the transgenes in brain neurons or photoreceptors.(9)
Transgenic Aß42 is severely neurotoxic to Drosophila, reducing lifespan from 38 days in controls to 28 d (WT Aß42) or 9 d (E22G). E22G neurotoxicity was also evident in the eye, where expression led to abnormal photoreceptor morphology. ZAβ3 substantially rescued fly lifespan against the expressed Aß, to 32 and 20 d, respectively. (ZAβ3)2 proved yet more effective: it fully rescued lifespan from the effects of WT Aß42 (to 40 d) and more fully normalized it against the effects of E22G (31 d), while substantially protecting photoreceptor morphology. Aß40 was harmless to the flies, and lifespan of TG-Aß40 Drosophila was unaffected by coexpression of affibodies.
Affibody clearance of Aß E22G was also evident, with Aß levels being high in E22G-TG Drosophila but considerably reduced in flies coexpressing ZAβ3 and indetectable with coexpression of (ZAβ3)2. That the mechanism of Aß reduction was not due to effects on Aß production was demonstrated by the lack of a significant effect on neuronal E22G expression. More intriguingly, the reduction in Aß was also not accompanied by a concomitant buildup in SDS-stable ZAβ3-Aß complexes, as demonstrated by a lack of appearance of liberated Aß following treatment with guanidinium chloride to dissociate such complexes. Rather, the absolute, total level of Aß in E22G-TG Drosophila was ~75% lower in flies coexpressing ZAβ3 monomer, and by 97±3% with coexpressed (ZAβ3)2, relative to levels in controls coexpressing inert Z domain.(9)
Reductions in monomeric Aß were accompanied by equivalent reductions in Aß fibrils in vivo, and in vitro studies showed that addition of affibody to Aß42 or -40 isoforms exerts equivalent effects on the kinetics of fibril formation as do equivalent reductions in initial levels of Aß monomer in the medium, implying that the reduction in fibril levels is primarily due to the initial binding of affibody to Aß monomer. Yet addition of excessive affibody later in the aggregation reaction process also arrests further fibril formation and even leads to a very gradual reduction in levels of existing fibrils, accompanied by the appearance of affibody-bound Aß monomers; this result suggests some ability to bind Aß monomers within fibrillar and protofibrillar Aß structures, and to slowly contribute to their dissociation (although at rates so low as to be unlikely to occur in situ in the brain).(9)
On the other hand, similar studies not only revealed a similar ability of ZAβ3 to prevent the formation of Aß oligomers from monomeric Aß, but also a physiologically plausible destabilization and dissolution of existing oligomers.(9)
The sum of the results suggest that initial sequestration of Aß monomers by ZAβ3 -- as free Aß molecules or as constituents of oligomeric and perhaps protofibrillar or even fibrillar Aß -- is later followed by degradation of affibody-bound Aß in vivo. The authors note that the mechanism underlying any such effect cannot involve clearance of Aß species by anti-Aß antibodies induced in the flies through activation of an adaptive immune response, as Drosophila do not have an adaptive immune system. Rather, they suggest that affibody-bound Aß monomers are subsequently degraded by endogenous biological mechanisms, such as proteasomal or lysosomal breakdown within neurons, or by phagocyes following an hypothetical exocytosis.(9) If correct, the rate of such degradation could in principle be enhanced if necessary by fortification of the lysosome with novel hydrolases (lysoSENS) targeting residual lysosomal Aß accumulations or any specific degradation-resistant aggregates formed following lysosomal uptake and storage.(13-15)
Obviously, these studies, while promising, are very preliminary. Drosophila is not an ideal model organism in which to test the effects of Aß and its clearance in vivo, and neuronal ZAβ33 coexpression studies can only be taken as suggestive of the possible effects of affibody delivered by systemic injection. Moreover, there remain many open questions and concerns about the timecourse and mechanism of Aß disappearance, such as whether ZAβ33 can indeed clear out existing accumulations of Aß oligomers and fibrils in aging human brains, and whether a mechanism apparently largely reliant on sequestration of free monomers might interfere with any putative physiologic role of Aß in the brain(10-12).
But a novel route toward Aß removal that does not rely on the immune system, and that offers the promise of removal of a wide range of Aß aggregates but especially of oligomeric species, is a promising new development in the field. Indeed, while speculation from this preliminary result cannot be given much weight, under a best-case scenario one can envision the opening of an entirely new approach to therapeutic clearance not only of Aß, but of a range of other disease- and aging-associated intracellular and extracellular aggregates. The broad outlines of the line of investigation required to turn these preliminary results into a therapy for brain aging and AD is reasonably clear; if its potential is realized clinically, then affibody-based therapeutics may ultimately be used to prevent and even reverse age-related diseases ranging from other forms of neurodegeneration, to senile cardiac amyloidosis, to age-related macular degeneration, to atherosclerosis. We await further revelations from these investigators with tempered optimism.
References
1: Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. Review. Erratum in: Nat Rev Neurol. 2010 Apr;6(4):183. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
2. Mamikonyan G, Necula M, Mkrtichyan M, Ghochikyan A, Petrushina I, Movsesyan N, Mina E, Kiyatkin A, Glabe CG, Cribbs DH, Agadjanyan MG. Anti-A beta 1-11 antibody binds to different beta-amyloid species, inhibits fibril formation, and disaggregates preformed fibrils but not the most toxic oligomers. J Biol Chem. 2007 Aug 3;282(31):22376-86. Epub 2007 Jun 1. PubMed PMID: 17545160; PubMed Central PMCID: PMC2435219.
3. Necula M, Breydo L, Milton S, Kayed R, van der Veer WE, Tone P, Glabe CG. Methylene blue inhibits amyloid Aß oligomerization by promoting fibrillization. Biochemistry. 2007 Jul 31;46(30):8850-60. Epub 2007 Jun 27. PubMed PMID: 17595112.
4. Necula M, Kayed R, Milton S, Glabe CG. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem. 2007 Apr 6;282(14):10311-24. Epub 2007 Feb 6. PubMed PMID: 17284452.
5. Wang MS, Boddapati S, Sierks MR. Antifibrillizing agents catalyze the formation of unstable intermediate aggregates of beta-amyloid. Biotechnol Prog. 2010 Feb 8. [Epub ahead of print] PubMed PMID: 20306540.
6. Petrushina I, Ghochikyan A, Mktrichyan M, Mamikonyan G, Movsesyan N, Davtyan H, Patel A, Head E, Cribbs DH, Agadjanyan MG. Alzheimer's disease peptide epitope vaccine reduces insoluble but not soluble/oligomeric Aß species in amyloid precursor protein transgenic mice. J Neurosci. 2007 Nov 14;27(46):12721-31. PubMed PMID: 18003852; PubMed Central PMCID: PMC2366938.
7. Nygren PA. Alternative binding proteins: affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 2008 Jun;275(11):2668-76. Epub 2008 Apr 24. Review. PubMed PMID: 18435759.
8. Grönwall C, Jonsson A, Lindström S, Gunneriusson E, Ståhl S, Herne N. Selection and characterization of Affibody ligands binding to Alzheimer amyloid beta peptides. J Biotechnol. 2007 Jan 30;128(1):162-83. Epub 2006 Sep 27. PubMed PMID: 17088007.
9. Luheshi LM, Hoyer W, de Barros TP, van Dijk Härd I, Brorsson AC, Macao B, Persson C, Crowther DC, Lomas DA, Ståhl S, Dobson CM, Härd T. Sequestration of the Aß peptide prevents toxicity and promotes degradation in vivo. PLoS Biol. 2010 Mar 16;8(3):e1000334. PubMed PMID: 20305716; PubMed Central PMCID: PMC2838747.
10. Morley JE, Farr SA, Banks WA, Johnson SN, Yamada KA, Xu L. A physiological role for amyloid-beta protein: enhancement of learning and memory. J Alzheimers Dis. 2010 Jan;19(2):441-9. PubMed PMID: 19749407.
11. Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009 Dec;12(12):1567-76. PubMed PMID: 19935655.
12. Pearson HA, Peers C. Physiological roles for amyloid beta peptides. J Physiol. 2006 Aug 15;575(Pt 1):5-10. Epub 2006 Jun 29. Review. PubMed PMID: 16809372; PubMed Central PMCID: PMC1819417.
13. de Grey AD, Alvarez PJ, Brady RO, Cuervo AM, Jerome WG, McCarty PL, Nixon RA, Rittmann BE, Sparrow JR. Medical bioremediation: prospects for the application of microbial catabolic diversity to aging and several major age-related diseases. Ageing Res Rev. 2005 Aug;4(3):315-38. PubMed: 16040282.
14. de Grey AD. Appropriating microbial catabolism: a proposal to treat and prevent neurodegeneration. Neurobiol Aging 2006;27(4):589-595. PubMed: 16207503.
15. de Grey AD. Lysosomal enhancement with microbial hydrolases: a novel strategy for removing protein aggregates. In: A. Fisher et al. (eds), New Trends in Alzheimer and Parkinson Disorders: ADPD 2005. Medimond, 2005: 51-4.
Posted 11 May 2010 - 02:02 AM
To date, all successful interventions into the biological aging process in experimental animals have entailed modulation of basic metabolic pathways, generally through genetic or dietary manipulation.(0) Of these, the earliest, most well-studied, and arguably the most robust, is Calorie restriction (CR): the reduction in dietary energy intake, without compromise of essential nutrients.(1,2) With few exceptions, CR retards the biological rate of aging in nearly every species and strain of organisms in which it has been tested, ranging from rotifers, through small multicellular invertebrates, and most extensively to laboratory rodents; and although inconclusive, recent evidence also supports its effectiveness in dogs(3) and nonhuman primates.(4) Moreover, while necessarily preliminary, a growing body of human research has reported that rigorous CR, when practiced by previously normal-weight adults, results in physiological, functional, and perhaps even structural changes consistent with its translation to the human case.(5-8)
While by no means universal (eg. (9,10)), there is therefore widespread optimism in the biogerontology community that CR would be similarly effective in humans. The most vocal such investigator was UCLA's Dr. Roy Walford, who was the first of small number of human prolongevists (of which the present author is one exemplar) to be sufficiently impressed by this evidence as to take up long-term, rigorous CR themselves, in hopes of enjoying the longevity and protection against age-related disease and disability observed in other species. Some members of the nonprofit CR Society, cofounded by Dr. Walford, are CR practitioners, and some of these are the subjects of one the most informative of the human CR studies.(eg, (5-8)).
This widespread optimism as to the translatability of CR, however, is tempered by equally widespread pessimism that any significant number of humans will take it up in practice. Instead, the focus of biogerontologists' thinking surrounding the potential clinical implications of CR research has long been to identify the core mechanism(s) responsible for the extension of youthful health and lifespan by CR, and to then target those mechanisms with "CR mimetics:" small molecules that would induce the "anti-aging" effects of CR, while neither requiring nor resulting in any reduction of energy intake. The CR mimetic concept was first formally formulated by Lane, Ingram, Roth of the National Institute on Aging (NIA) in 1998(11) and has been a subject of growing interest ever since (eg, (2,12)).
But despite the initial attractiveness of the notion; its strong theoretical basis; the high level of scientific interest that it has garnered; the launching of biotech startups originating in CR mimetic research; and the popularization and commercial exploitation of the concept by the dietary supplement industry -- despite all of these drivers, the ensuing decade and a half or more of CR mimetic research have thus far been fruitless. Initially-promising compounds have failed to extend lifespan, while surprising findings have preempted the further investigation of what might otherwise have been novel targets for CR mimetics. Here we review some of the more prominent cases.
Antioxidants -- Free Radicals. The free radical theory of aging -- both in its early, simplistic form, and in its later and more refined iterations -- and the support afforded to it by the CR data, will be familiar to readers of this forum, and need not detain us. The same is likely true of intervention with dietary antioxidants, which have repeatedly been the subject of testing in animal models and in human clinical trials, and which have repeatedly failed to (respectively) extend lifespan in normal, healthy mammals, or to improve clinical outcomes in humans at risk for disease. These elementary facts bear brief mention for completeness; having made such allusion, we will now move on to less well-known disappointments.
2-deoxyglucose (2DG) -- Nutrient Sensing. This was the first agent to be formally investigated as a putative CR mimetic; indeed, the initial report of these studies marked the first formulation of the concept and the coining of the term "CR mimetic" itself.(11) 2DG, an unmetabolizable analog of glucose, exhibits similar pharmacokinetics as the parent molecule, including in its cellular uptake and initial entry into the glycolytic pathway -- properties that had previously been exploited to probe glycolysis itself, and as a radioactive tracer for PET scanning. Following its initial metabolism by hexokinase, further metabolism of 2DG is arrested, and it therefore acts as a competitive inhibitor of cellular glucose metabolism. Lane et al's interest in this compound were based on prior reports indicating that 2DG induced physiological and bioenergetic effects parallel to CR, including lowering body temperature, altering reproductive function, and even inhibiting tumor growth. Their own initial studies showed that 2DG feeding also lowered serum glucose and insulin levels in rats, while causing only relatively minor reductions in body weight;(11) later studies by their group and others found that 2DG lowers heart rate, elevates circulating glucocorticoids and expression of heat shock proteins (effects thought to underlie a protective "hormetic" mechanism of CR), increases resistance to cold shock, affordins substantial protection against ischaemic and toxic insults to the brain, and increases mean and apparently maximal lifespan in Caenorhabditis elegans (reviewed in (14)).
But mammalian lifespan studies of 2DG proved initially frustrating, and ultimately futile. A first study at doses that had induced a CR-like metabolic pattern in rats led to premature deaths associated with congestive heart failure and cardiac vacuolization, while lower dosages sufficient to avoid cardiac toxicity failed to alter metabolism and had no effect on lifespan.((11-14); Figure 1). Later studies using alternative material sources (to rule out a possible role for contaminants) and different strains of rat confirmed the compound's cardiotoxicity, with histopathology additionally finding pheochromocytoma in 2-DG-fed rats.
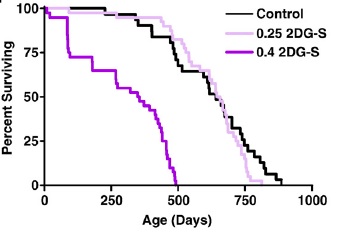
Figure 1. Survival of F344 rats on 2 doses of 2-deoxyglucose. Reproduced from (14).
Metformin -- Insulin Signaling, AMPK, Gene Expression. CR robustly increases insulin sensitivity, lowers blood glucose, and reduces the risk of diabetes in rodents and nonhuman primates, and of impaired glucose tolerance in humans. These effects have long been postulated to be central to the retardation of aging by CR.(15) Metformin, an hepatic gluconeogenesis inhibitor and mild insulin sensitizer used in the treatment of diabetes, emerged as a widely-cited possible CR mimetic as a result of these effects, along with its activation of AMPK, the wide overlap of its effects on hepatic gene expression profiles with those of CR, and a possible reduction in mitochondrial free radical generation due to inhibition of Complex I of the electron transport chain.(16) These mechanistic findings were bolstered by the protective effects of related biguanide drugs in genetic models of carcinogenesis; by the reduction of mortality in a short-lived mice when administered metformin itself; and by the uniquely protective effects of metformin (against total and cancer mortality observed in diabetic patients.(16) But as reviewed elsewhere, the long-delayed results of a careful life extension study using metformin in F344 rats have recently published, finding no effect -- although there was sufficient ambiguity in the results the issue cannot yet be said to be fully resolved.(17) Separate studies on metformin are underway in the lab of Dr. Steven Spindler, UC Riverside.

Figure 2. Kaplan–Meier survival plots for CON, CR, MET, and PF–MET (CON: n = 31; CR: n = 40; MET; n = 40; and PF–MET: n = 40). Reproduced from (17).
Pimagedine -- Glycation and Advanced Glycation Endproducts. Along with its reduction of circulating glucose and triglycerides, CR has repeatedly been found to reduce the age-related accumulation of advanced glycation endproducts (AGE) in the tissues of animals. Due to the implication of AGE in the complications of diabetes and the age-related tissue dysfunction, this structural effect of the altered metabolism of fuels by CR has been specifically highlighted as a potentially central mechanism of CR.(16) The rate of accumulation of the markers of glycation and glycoxidation in tissue collagen was the only promising candidate biomarker of aging to emerge in relation to the NIA's Biomarkers of Aging program, and was specifically found to be a limiting factor for the individual survival of both CR and ad libitum mice. (18) In two studies unfortunately marred by animal cohorts with historically low longevity, the length of life was greater when fed a low-AGE chow,(19) while high-AGE chow blunted of the extension of life by CR.(20)
Pimagedine (aminoguanidine), a drug once under investigation as a therapeutic for the complications of diabetes, is a competitive inhibitor of AGE formation, sequestering reactive dicarbonyl intermediates and retarding the rate of accumulation of AGE in the tissues of rodents, and was the subject of much interest as a possible longevity therapeutic and partial CR mimetic. However, it failed to meet its primary outcome in clinical trials in diabetic subjects (creatinine doubling time) and was associated with a range of mild to moderate adverse reaction,(21) and rodent studies carried out by Dr. Spindler found no benefit in normal, nondiabetic mice.(16)

Figure 3. Survival of mice administered control diet alone (solid black square), or with aminoguanidine (downward pointing triangle; 65 mg/kg body weight/d), aminoguanidine and alpha-lipoic acid (hollow diamond; 65 and 73 mg/kg. respectively), aminoguanidine, alpha-lipoic acid, pregnenolone, and coenzyme-Q 10 (hollow circle; 65, 73, 0.2, and 12 mg/kg), melatonin (hollow square, 41 µg/kg), or melatonin and pregnenolone (upward pointing triangle, 41 and 200 µg/kg). Reproduced from (16).
Resveratrol -- Sirtuins. By far the most well-publicized possible CR mimetic ever was the phytoalexin polyphenol resveratrol, present in trace amounts in grapes and (famously) wine. Interest in resveratrol was initially sparked by research on Sir2, an NAD+-dependent histone deacetylase in the baker's yeast Saccharomyces cerevisiae. Reports in S. cerevisiae, C. elegans, and Drosophila melanogaster indicated that lifespan could be extended in these organisms by 30–50% by increased copy number or expression of the gene; because its activity was reponsive to the cellular NAD+:NADH ratio, and because of reports that life extension by CR-like dietary manipulation in S. cerevisiae and Drosophila required Sir2, it was hyypothesized that Sir2 activation might be a key mechanism of CR. Investigation of resveratrol as a CR mimetic began with a report that it was one of a small number of "sirtuin activating compounds (STAC)" identified in a screen using recombinant human SIRT1 (the human homolog) in vitro, and the hypothes was underscored by reports that administration of resveratrol extended life in the same range of lower organisms as had already been reported to respond to Sir2. Nearly all of these claims were later disputed,(22-27) but significant public and scientific interest had already been generated, and experiments in mice were initiated.
Interest increased dramatically by reports that high-dose resveratrol supplements partially normalized lifespan(28) and various aspects of health and functionality(28,29) in mice made obese and diabetic by a high-hydrogenated-coconut-oil diet. In the popular press, to a limited degree in the scientific literature, and especially in dietary supplement companies' promotional materials, these results were often misconstrued as demonstrating actual extension of normal, youthful functionality and lifespan, leading to remarkably widespread interest and enthusiasm.
But finally, in 2008, the results of a lifespan study of 3 doses of resveratrol in normal, healthy mice were published.(30) While some specific aspects of age-related deterioration were retarded in resveratrol-fed mice, survival and pathology were unaffected. Surprisingly, this negative result has had virtually no effect on media coverage, and mention of the result is (unsurprisingly) studiously avoided in promotional material.

Figure 4. Effects of resveratrol on mice fed standard diet (SD) or SD plus a low (100 mg/kg food = 7.9 ± 0.2 mg/kg body weight, SDLR) or high (400 mg/kg food = 30.9 ± 0.6 mg/kg wt) dose of resveratrol. "Later, additional groups of mice were given a higher dose of resveratrol along with the standard ... diets (2400 mg/kg of food, SDHR) ... beginning at 12 months of age (SDHR) on lifespan, and again found that longevity was not significantly affected." Reproduced from (30).
Additional studies are underway to test resveratrol at several doses and with 2 ages of initiation through the NIA's Interventions Testing Program, "a multi-institutional study investigating treatments with the potential to extend lifespan and delay disease and dysfunction in mice."
Rapamycin -- mTOR. Finally, however, 2009 saw the publication of a successful CR mimetic. Rapamycin is an inhibitor of the mammalian Target of Rapamycin (mTOR), a highly conserved protein kinase serine/threonine kinase that integrate signals from nutrients (cellular ATP and amino acid levels) and trophic signaling (insulin/insulin-like growth factor 1 (IGF-1) and other mitogens) to regulate autophagy, cell growth, and cell cycle progression. Rapamycin (sirolimus /Rapamune®) and a range of analogs are therefore already FDA approved cancer treatments, immunosuppressants, and cardiology drugs, and additional analogs are in development.
Inhibition of TOR has emerged in recent years as a strong candidate mechanism of CR, due to its involvement in cellular response to energetic and trophic signaling, its inhibition during CR in a wide range of animal models, and the extension of life in several such models garnered by disrupting its signaling pathway.(31) It was therefore put into testing in the NIA's ITP. And after a series of disappointments with other modulators of putative metabolic mediators of biological aging, these studies led the programto an historic first. Rapamycin is now the first pharmaceutical intervention to robustly extend the normal lifespan of genetically intact, well-husbanded mammals (genetically heterotgeneous mice) of both genders.(32) Even more impressively, it was shown to be effective when administered relatively late in life (600 d, vs. historical and cohort control mean and maximum lifespans of ~900 d and 1100-1200 d, respectively, for mice).
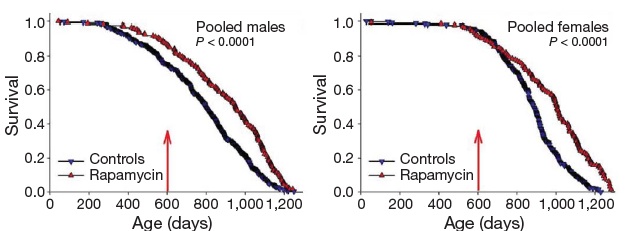
Figure 5. Effect of late-life rapamycin administration on survival in mice. Reproduced from (32)
The result being rightly heralded for the landmark advance in biogerontology it undoubtedly is (it received an honorary Mprize Lifespan Achievement Award), it none the less illustrates the limitations of the CR mimetic approach. While the relative gain in mean lifespan (remaining life expectancy at first exposure to rapamycin (600 d)) was substantial (28% for males, 38% for females), the absolute gains were relatively modest: 9% and 13%, respectively (and similarly for tenth-decile survivorship).(32) CR itself, when implemented in longevous male at a similar age, has been reported to increase maximum longevity by ~16%(33).
These gains are enormous, when compared to the multiple null results or even harmful effects reported in previous studies, they are in absolute terms relatively small -- and certainly smaller than the ~40% increases in mean and maximal lifespan gained when CR is initiated shortly after weaning. And it is to these latter, far higher figures that advocates of the CR mimetic approach often refer when making their case (eg. 34):
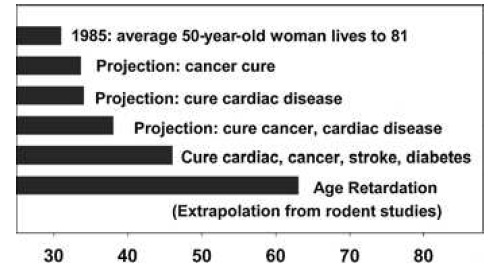
Figure 6. "Remaining life expectancy of a 50-year-old Caucasian woman in the United States in 1985, at then-current mortality risk schedule (top bar), or as projected under the assumption that adult mortality risks for specific diseases (cancer, cardiac disease, etc., as indicated) were reduced to zero from 1985 onward. The bottom bar shows projected life expectancy if human adult mortality risks could be reduced to the same extent that caloric restriction reduces them in mice." Legend and figure reproduced from (34).
... despite the obvious fact that there is no prospect, for ethical reasons, of such an intervention protocol ever being carried out in humans. It is rather in persons of middle age and above that even the most liberal of regulatory and oversight bodies would allow a CR mimetic to be administered, supposing them to already be in reasonable good health for their age. It is thus to the more modest figures for late-life intervention (whether by CR or proper) that reference must be made to make a case that accurately reflects the underlying data.
The support lent the CR mimetic approach by the rapamycin result must also be tempered by a comparison of the results of this study (32) to the effects of CR itself. While one must be cautious in comparing results acquired using different protocols and in different laboratories, it is fully as one would expect that the lifespan gains reported for rapamycin are significantly less than those reported for CR in the same gender and initiated at the same age (9% vs ~16%). The more modest reported effect of rapamycin has several plausible and somewhat trivial explanations, which are not mutually exclusive, including differences in rodent strain, husbandry, and other protocols, and especially the optimization of the intervention dose. But one that should be highlighted, and would be expected to apply on a priori grounds to even the most rigorously-matched set of experimental conditions, is that it is unreasonable to expect that a pharmacological mimetic of CR will be as effective as CR itself. Leaving aside the potential limitation of the benefits of any drug therapy by off-target effects, it seems unlikely that all of the pleiotropic effects of CR but one (such as inhibition of mTOR signaling) are dispensable to its ability to extend the healthy, youthful lifespan.
Is it reasonable to expect, for example, that the full contribution to the retardation of biological aging by CR afforded by reductions in insulin, IGF-1, cellular amino acids, and ATP are mediated by downstream inhibition of mTOR? Is it not more plausible to expect that these effects of CR contribute to the modulation of aging through additional, unrelated mechanisms? And what of the many physiological effects of CR that do not affect the mTOR pathway at all, such as the reductions circulating glucose, lipids, and lipoproteins, body temperature, sex steroids, and visceral adiposity? Are we to think that the modulation of these multiple disease- and aging-associated parameters by CR is fully dispensible to the extension of youth, supposing only that mTOR activity is optimally suppressed? Rather, it seems more reasonable to expect that the only intervention that will fully deliver the sought-for gains in healthy lifespan derived from CR, will be CR itself.* All pharmacological mimics will, in this analysis, be expected to be shadows of CR itself, of greater or lesser opacity.
Beyond CR Mimetics. Recognizing the inherent limitations of the CR mimetic concept as a strategy to develop therapeutic interventions against degeneration of biological aging, regenerative engineering is advanced as an alternative. Rather than attempting to modulate basic metabolic pathways in hopes of reducing the deleterious side-effects that they produce, regenerative engineering proposes to develop a new class of therapeutics that directly remove, repair, replace, or render harmless the cellular and molecular damage that accumulates in tissues over time, impairing functionality and resulting in the progressive rise in frailty, disease, disability, and death that people now suffer with advancing age. With the burden of such damage removed from aging tissues, their structure and in principle functionality would be restored, leading to the renewal of youthful health and vigor. And because this strategy does not necessitate the modulation of metabolic pathways to be effective, regenerative engineering therapeutics should be less prone to generating the deleterious side-effects that must inevitably accompany interference with the biochemical basis of life itself. SENS Foundation is dedicated to accelerating the development of a comprehensive suite of regenerative engineering interventions, to prevent and even reverse the degenerative aging process in as many persons as possible, on the most aggressive possible schedule.
References
* On the other hand, it also bears mention that it is equally unreasonable to think that all of the physiological effects of CR are necessary contributors to its benefits. If so, then a CR mimetic that bypassed some such effects might deliver (some of) the benefits of CR while dispensing with (some of) its undeniable deleterious or troublesome effects.(35)
0. Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008 Aug 28;454(7208):1065-71. Review. PubMed PMID: 18756247; PubMed Central PMCID: PMC2774752.
1. Weindruch R., Walford R. L. The Retardation of Aging and Disease by Dietary Restriction. 1988; Charles C. Thomas Springfield, IL.
2. Smith DL Jr, Nagy TR, Allison DB. Calorie restriction: what recent results suggest for the future of ageing Research. Eur J Clin Invest 2010; 40 (5): 440-50.
3. Kealy RD, Lawler DF, Ballam JM, Mantz SL, Biery DN, Greeley EH, Lust G, Segre M, Smith GK, Stowe HD. Effects of diet restriction on life span and age-related changes in dogs. J Am Vet Med Assoc. 2002 May 1;220(9):1315-20. PubMed PMID: 11991408.
4. Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009 Jul 10;325(5937):201-4. PubMed PMID: 19590001; PubMed Central PMCID: PMC2812811.
5. Cangemi R, Friedmann AJ, Holloszy JO, Fontana L. Long-term effects of calorie restriction on serum sex-hormone concentrations in men. Aging Cell. 2010 Apr;9(2):236-42. Epub 2010 Jan 20. PubMed PMID: 20096034.
6. Fontana L, Klein S, Holloszy JO. Effects of long-term calorie restriction and endurance exercise on glucose tolerance, insulin action, and adipokine production. Age (Dordr). 2010 Mar;32(1):97-108. Epub 2009 Nov 11. PubMed PMID: 19904628; PubMed Central PMCID: PMC2829643.
7. Fontana L, Weiss EP, Villareal DT, Klein S, Holloszy JO. Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentration in humans. Aging Cell. 2008 Oct;7(5):681-7. PubMed PMID: 18843793; PubMed Central PMCID: PMC2673798.
8. Holloszy JO, Fontana L. Caloric restriction in humans. Exp Gerontol. 2007 Aug;42(8):709-12. Epub 2007 Mar 31. Review. PubMed PMID: 17482403; PubMed Central PMCID: PMC2020845.
9. de Grey AD. The unfortunate influence of the weather on the rate of ageing: why human caloric restriction or its emulation may only extend life expectancy by 2-3 years. Gerontology. 2005 Mar-Apr;51(2):73-82. Review. PubMed PMID: 15711074.
10. Shanley DP, Kirkwood TB. Caloric restriction does not enhance longevity in all species and is unlikely to do so in humans. Biogerontology. 2006 Jun;7(3):165-8. PubMed PMID: 16858629.
11. Lane MA, Ingram DK, Roth GS. 2-Deoxy-D-glucose feeding in rats mimics physiologic effects of calorie restriction. J Anti-Aging Med. 1998 Winter;1(4):327-37.
12. Minor RK, Allard JS, Younts CM, Ward TM, de Cabo R. Dietary Interventions to Extend Life Span and Health Span Based on Calorie Restriction. J Gerontol A Biol Sci Med Sci. 2010 Apr 6. [Epub ahead of print] PubMed PMID: 20371545.
13. Lane MA, Mattison J, Ingram DK, Roth GS. Caloric restriction and aging in primates: Relevance to humans and possible CR mimetics. Microsc Res Tech. 2002 Nov 15;59(4):335-8. Review. PubMed PMID: 12424798.
14. Minor RK, Smith DL Jr, Sossong AM, Kaushik S, Poosala S, Spangler EL, Roth GS, Lane M, Allison DB, de Cabo R, Ingram DK, Mattison JA. Chronic ingestion of 2-deoxy-D-glucose induces cardiac vacuolization and increases mortality in rats. Toxicol Appl Pharmacol. 2010 Mar 15;243(3):332-9. Epub 2009 Dec 22. PubMed PMID: 20026095; PubMed Central PMCID: PMC2830378.
15. Masoro EJ, McCarter RJ, Katz MS, McMahan CA. Dietary restriction alters characteristics of glucose fuel use. J Gerontol. 1992 Nov;47(6):B202-8. Erratum in: J Gerontol 1993 Mar;48(2):B73. PubMed PMID: 1430849.
16. Spindler SR, Mote PL. Screening candidate longevity therapeutics using gene-expression arrays. Gerontology. 2007;53(5):306-21. Epub 2007 Jun 15. Review. PubMed PMID: 17570924.
17. Smith DL Jr, Elam CF Jr, Mattison JA, Lane MA, Roth GS, Ingram DK, Allison DB. Metformin Supplementation and Life Span in Fischer-344 Rats. J Gerontol A Biol Sci Med Sci. 2010 Mar 19. [Epub ahead of print] PubMed PMID: 20304770.
18. Sell DR, Kleinman NR, Monnier VM. Longitudinal determination of skin collagen glycation and glycoxidation rates predicts early death in C57BL/6NNIA mice. FASEB J. 2000 Jan;14(1):145-56. PubMed PMID: 10627289
19. Cai W, He JC, Zhu L, Chen X, Wallenstein S, Striker GE, Vlassara H. Reduced oxidant stress and extended lifespan in mice exposed to a low glycotoxin diet: association with increased AGER1 expression. Am J Pathol. 2007 Jun;170(6):1893-902. PubMed PMID: 17525257; PubMed Central PMCID: PMC1899464.
20. Cai W, He JC, Zhu L, Chen X, Zheng F, Striker GE, Vlassara H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am J Pathol. 2008 Aug;173(2):327-36. Epub 2008 Jul 3. PubMed PMID: 18599606; PubMed Central PMCID: PMC2475771.
21. Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, Foiles PG, Freedman BI, Raskin P, Ratner RE, Spinowitz BS, Whittier FC, Wuerth JP; ACTION I Investigator Group. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004 Jan-Feb;24(1):32-40. Epub 2003 Dec 17. PubMed PMID: 14685005
22. Garber K. A mid-life crisis for aging theory. Nat Biotechnol. 2008 Apr;26(4):371-4. PubMed PMID: 18392009.
23. Ledford H. Ageing: Much ado about ageing. Nature. 2010 Mar 25;464(7288):480-1.
24. Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell S, Napper A, Curtis R, Distefano PS, Fields S, Bedalov A, Kennedy BK. Substrate specific activation fo sirtuins by resveratrol. J Biol Chem. 2005 Jan 31; [Epub ahead of print] PMID: 15684413
25. Zou S, Carey JR, Liedo P, Ingram DK, Müller HG, Wang JL, Yao F, Yu B, Zhou A. The prolongevity effect of resveratrol depends on dietary composition and calorie intake in a tephritid fruit fly. Exp Gerontol. 2009 Jun-Jul;44(6-7):472-6. Epub 2009 Mar 3. PubMed PMID: 19264118.
26. Bass TM, Weinkove D, Houthoofd K, Gems D, Partridge L. Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech Ageing Dev. 2007 Oct;128(10):546-52. Epub 2007 Aug 14. PubMed PMID: 17875315.
27. Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, Qiu X, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010 Mar 12;285(11):8340-51. Epub 2010 Jan 8. PubMed PMID: 20061378; PubMed Central PMCID: PMC2832984.
28. Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006 Nov 16;444(7117):337-42. Epub 2006 Nov 1. PMID: 17086191 [PubMed - indexed for MEDLINE]
29. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006 Dec 15;127(6):1109-22. Epub 2006 Nov 16. PMID: 17112576 [PubMed - indexed for MEDLINE]
30. Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, Jamieson HA, Zhang Y, Dunn SR, Sharma K, Pleshko N, Woollett LA, Csiszar A, Ikeno Y, Le Couteur D, Elliott PJ, Becker KG, Navas P, Ingram DK, Wolf NS, Ungvari Z, Sinclair DA, de Cabo R. Resveratrol Delays Age-Related Deterioration and Mimics Transcriptional Aspects of Dietary Restriction without Extending Life Span. Cell Metab. 2008 Aug;8(2):157-68. PMID: 18599363 [PubMed - as supplied by publisher]
31. Stanfel MN, Shamieh LS, Kaeberlein M, Kennedy BK. The TOR pathway comes of age. Biochim Biophys Acta. 2009 Oct;1790(10):1067-74. Epub 2009 Jun 16. Review. PubMed PMID: 19539012.
32. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009 Jul 16;460(7253):392-5. Epub 2009 Jul 8. PubMed PMID: 19587680; PubMed Central PMCID: PMC2786175.
33. Dhahbi JM, Kim HJ, Mote PL, Beaver RJ, Spindler SR. Temporal linkage between the phenotypic and genomic responses to caloric restriction. Proc Natl Acad Sci U S A. 2004 Apr 13;101(15):5524-9. Epub 2004 Mar 25. PubMed PMID: 15044709; PubMed Central PMCID: PMC397416.
34. Miller RA. Extending life: scientific prospects and political obstacles. Milbank Q. 2002;80(1):155-74. PMID: 11933792 [PubMed - indexed for MEDLINE]
35. Dirks AJ, Leeuwenburgh C. Caloric restriction in humans: potential pitfalls and health concerns. Mech Ageing Dev. 2006 Jan;127(1):1-7. Epub 2005 Oct 13. Review. PubMed PMID: 16226298.
Posted 06 May 2010 - 11:25 PM
Johan Svantesson Sjöberg is in his second year at Lund University, pursuing a BS in Molecular Biology. He has worked with the SENSFAI team since 2008, and is currently a volunteer on the Project Development Committee.
<!--break--> His first project with SENSFAI was working with a partner to write a review paper on the characteristics, formation and pathophysiology of the cross-link glucosepane. After more than half a year of work, their project was published in the peer-reviewed journal "Rejuvenation Research" in April or 2009.
His first project with SENSFAI was working with a partner to write a review paper on the characteristics, formation and pathophysiology of the cross-link glucosepane. After more than half a year of work, their project was published in the peer-reviewed journal "Rejuvenation Research" in April or 2009.
"I think it's really exciting that, in the near future, we have a chance of being able to do something significant about problems like aging, when I found out about the masters programme in bioinformatics at Lund University, I thought it would be a good way to combine molecular biology with my interest in computers."
You can read the text of our interview below:
Currently, I have completed the first year (of three) of a B.Sc. in molecular biology. After that, I'm planning on studying for a two-year M.Sc. in bioinformatics, and then I would like to continue with Ph.D. studies, hopefully related to protein modelling or something similar. Neither of my parents were academics, so this is a journey into a new and exciting world for me. So far I'm really enjoying it!
My interest for life extension research was awakened during my second year of gymnasium (Swedish equivalent to high school, three years). At the time, I was studying arts to become an architect. I have always been interested in science, but it wasn't until then that I started considering it as a career path. So I took some extra courses during my third year of gymnasium and attended a preparatory year at the college of Borås, and after that I was ready for university studies.
My long-term plan is probably to stay at Lund University as a researcher, and perhaps teaching as a lector or (eventually) professor. During the two years I've spent at Lund University so far, I've fallen in love with it, and I could very well imagine staying here for the rest of my career. I'm open to suggestions, though - for example, if I happen to come across an innovative idea during my research, I might start a company based on it.
After learning about life extension research and the Immortality Institute, in 2007 I found out about the Methuselah Foundation, as it was called then. My first involvement with the SENSFAI (named MFURI back then) was in 2008, when I and Sven Bulterijs did a project on the cross-link glucosepane (see next question below). After that, I have been working as a SENSFAI volunteer, writing project proposals for student projects.
An important part of what motivates me is personal interest, since I feel that the SENS strategy is the most promising way to combat the aging process. Another source of motivation is the fact that my involvement with SENSFAI is probably good for my career since, among other things, it allows me to gain experience with things like writing projects and publishing papers. SENSFAI is also a good way to meet great people and make new friends, and that's never a bad thing!
My current involvement with SENSFAI is volunteer work. I'm in the Project Development Committee which, as the name suggests, develops projects for students to work with. I have an idea for a lab project that I might be able to work with this summer, assuming I have enough time to write a project and grant proposal soon.
Yes, definitely - partly because it allows me to gain experience with writing projects and publishing papers, and partly because I can establish contacts with active researchers within the field of life extension.
Currently I am president of the student union of science (called "Luna") at Lund University. As president of Luna, I am representing the science students of Lund University in discussions and decisions within the Faculty of Science, e.g. by being in the faculty board, presidium, and Council of Education. I also organize and keep track of all of Luna's student volunteers.
I have a number of other positions within Luna as well, and after my term of office ends in July this year I will continue to be involved with Luna for the rest of my studies at Lund University. I think student unions fulfill a very important role in voicing the students' opinions in discussions with their respective universities. It's also a good way for me to gain personal contacts with a lot of professors and other university staff around here - might be good for the future, you never know...
Posted 30 July 2010 - 12:18 AM
Haematopoietic stem cells (HSC) and their progeny from exhibit a range of functional declines during biological aging. Most research probing the reasons for these declines have focused on aging damage accumulating in the HSCs themselves, such as the rising burden of oxidative stress and DNA damage (and, as a result, senescent cells) in the compartment. But there has been comparatively little exploration of the possibility of outside causes for age-related HSC dysfunctions, such as the role of age-related shifts in the systemic and local environment and the aging of the bone marrow HSC niche. In a recent report, Dr. Amy Wagers' stem cell group at Harvard and affiliated institutes1 have demonstrated the reality -- and the reversibility -- of both of these influences on age-related HSC dysfunction, using an intriguing model of systemic rejuvenation.
The investigators used the heterochronic rodent parabiosis model, in which the circulatory systems of young (2 mo old) and biologically aged (>20 mo) mice are surgically conjoined, allowing their circulatory systems to commingle and equilibrate; as controls, these young-to-old pairs are compared with age-matched pairs of each age group. In an earlier collaboration with Dr. Irena Conboy, Wagers had previously used this captivating experimental system to strikingly demonstrate the rejuvenating effects of exposure to a more youthful systemic environment on skeletal muscle satellite cell mobilization, proliferation, and regenerative capacity, and on the proliferation of hepatic progenitor cells.7For these new studies in HSC niche function, all parabiotic pairs were composed of partners that differed at the CD45 locus (CD45.1 and CD45.2), allowing haematopoietic cells to be traced to their originating hosts.
Youthful Circulation Restores Youthful Regulation of HSCs in Aged Animals
Comparing isochronic pairs from the 2 age groups, old mice exhibited superfluous accumulations of primitive long-term reconstituting HSCs relative to young comparitors. These cells were characterized by impaired haematopoietic engraftment, manifested in a less efficient reconstitution of peripheral blood leukocytes, and a bias toward the myeloid lineage at the expense of B-cell development. But all of these changes in old-derived HSC numbers and function were substantially normalized following exposure to the more youthful heterochronic environment (see Figure 1). The frequency and number of bone-forming osteoblasts in aged mice -- a constituent of the HSC niche with a central role in regulating HSC number and activity -- were also inflated (up to fourfold) in aged mice. Consistent with the findings in the parabiosis model, these old cells induced the development of more HSCs from cocultured lineage-negative bone marrow cells in vitro than did young-derived bone-forming osteoblasts, possibly explaining the age-related increase in long-term reconstituting HSCs formed in aged animals in vivo.1
Figure 1. Exposure of aged mice to youthful circulatory system restores long-term reconstituting HSC number and function and osteoblastic niche cell number. a: Frequency of endogenous LT-HSCs. b: Frequency of osteoblastic niche cells. c: Reconstitution of haematopoietic system in irradiated recipients by donor bone marrow cells 12 wk posttransplant. From (1). Rejuvenating Effects of Young Circulation is Mediated by the HSC Niche The fact that aged animals' osteoblastic HSC niche cells recapitulate the bias toward excessive accumulation of long-term reconstituting HSC observed in vivo in aged animals suggested that that the effects of a 'younger' heterochronic parabiotic systemic environment on old animals' HSC were the result of an indirect effect, mediated by a rejuvenated HSC niche. Indeed, exposure to a more youthful systemic environment through heterochronic parabiosis normalized the old animals' frequency and number of bone-forming osteoblasts, and their rate of formation of long-term reconstituting HSCs from lineage-negative bone marrow cells ex vivo.1
More dramatically, lineage-neutral HSCs cocultured with old-derived osteoblastic niche cells demonstrated impaired engraftment and differentiation when transplanted into old or even young irradiated hosts, recapitulating the defects seen in old animals and reinforcing the importance of the aging of the niche in those defects in the aged host. In contrast, there was no effect of osteoblastic niche cells from aged animals on the engraftment, lineage bias, or reconstituting ability of HSCs from young animals.1
Role of Excess IGF-1 in Impaired HSC Niche Regulatory Function
Exposure of young-derived HSCs to serum derived from aged murine or human donors again led to superfluous long-term reconstituting HSC accumulation, and exposure of aged osteoblastic niche cells to young serum blunted their dysregulatory influence on young-derived cells. Exposure of such cells to aged niche cells also increased their expression of several age-related myeloid-biasing markers, and decreased their expression of lymphoid markers whose expression is known to be reduced in aged organisms, consistent with the observed effects of such cells on lineage in vivo. Notably, these shifts were not observed following coculture with niche cells derived from aged animals that had benefited from the rejuvenating systemic environment of heterochronic parabiosis.1
Further studies to probe the mechanistic basis of the systemic influence on the HSC niche led to the surprising conclusion that a significant mediator of the impaired HSC regulation of the aged osteoblastic HSC niche is an excess of local IGF-1 signaling. Prior exposure of old-derived haematopoietic osteoblastic niche cells or serum to anti-IGF-1antibodies abolished the abnormal accumulation of long-term reconstituting HSCs during coculture with young-derived HSCs. By contrast, anti-IGF-1 Abs had no effect on the influence of young niche on HSCs, nor on old-derived HSCs in isolation. Similar effects were observed on a dose-dependent basis in vivo following neutralizing antibody injection into aged animals' bone marrows, but not following systemic injection via the peritoneum, showing that the disrupting effect of excessive IGF-1 on HSC function occurs in the haematopoietic niche microenvironment, rather than in the circulation at large, narrowing the effects observed in circulatory parabiosis.1
This counterintuitive finding is superficially opposite to the effects of local IGF-1 expression observed in aging muscle.2 It is instructive, if only by analogy, to compare the contrasting pro- and anti-aging local effects of IGF-1 in aging organisms to the paradoxical findings in other systems, such as the restoration of declining IGF-1 in normally-aging organisms vs. the many models of retarded biological aging in mice and other species characterized by reduced IGF-1 signaling.3 Notably, the effects of these experimental systems on local IGF-1 signaling is in at least some cases tissue-specific: local brain IGF-1 levels and signaling are preserved or enhanced in several models of retarded aging characterized by low systemic IGF-1 levels.4-6 And while systemic IGF-1 levels are low throughout the lifespan in these models, they remain stable at advanced ages when they have declined substantially in normally-aging animals; moreover, Calorie restriction preserves the regulated, pulsatile release of growth hormone with aging (Fig. 2) and selectively retains celllular IGF-1 signaling, even as these capacities are progressively lost in animals fed ad libitum.17-19
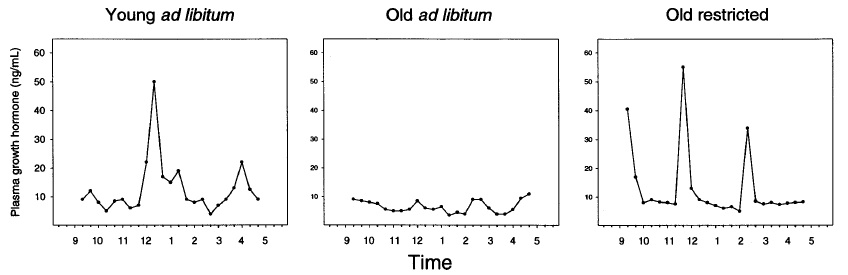
Figure 2. Preservation of growth hormone secretory dynamics in by CR in aging Brown Norway rats. From (18)
Implications for Regenerative Engineering and WILT
Whole-body Interdiction of Lengthening of Telomeres (WILT, or OncoSENS) is proposed by de Grey et al(14,15) as an impregnable defense against cancer, as part of a comprehensive panel of regenerative engineering therapies to repair the damage and diseases of aging. At its core, it entails the ablation of gene(s) essential to the telomere maintenance machinery (TMM), accompanied by periodic re-seeding of somatic stem-cell pools with autologous cells rendered equally defective for telomere elongation but whose telomeres have been lengthened ex vivo to allow for ongoing tissue repair and maintenance. The strongest challenge to this approach has been the possible existence of functions of TMM other than the lengthening of telomeres itself,8-13 and that even if TMM were dispensible in telomere-elongated stem cells, it might be essential to the functioning of the niche.
SENS Foundation is now funding research by Dr. Zhenyu Ju (formerly a telomerase researcher in Dr. K. Lenhard Rudolph’s laboratory and now at the Max Planck Partner Group on Stem Cell Aging at the Chinese Academy of Medical Sciences) to help resolve the latter question, by monitoring the effects of transplanting telomerase-deficient but ex vivo telomere-extended bone marrow into first normal and then TMM-deficient mice. The finding that the age-related loss of HSC function is substantially attributable to derangement of HSC regulation by the aging niche, much of which is secondary to shifts in systemic factors in the aging niche microenvironment rather than to cell-autonomous defects, provides some very preliminary reassurance on this issue.
More confidently, this latest example of the rejuvenating effect of youthful systemic environment (cf. (7), and Conboy's later reports in rejuvenation of muscle satellite cell function, as well as loosely-related studies in rejuvenation of the aging female reproductive system) reinforces the expectation that the effects of regenerative engineering therapies will not be narrowly confined to restoring the function of their specific target tissues. As we remove, repair, replace, or render harmless the cellular and molecular damage of aging, the progressive restoration of normal cell and tissue function can be expected to result in a concomitant, progressive normalization of the systemic milieu, as oxidative stress, inflammation, endocrine and paracrine signaling, and other systemic responses to -- and sequelae of-- the damage of aging are obviated and the body's inherent maintenance capacities are engaged. With this normalization, these studies suggest, the deranging effects of an aged systemic environment will gradually be alleviated, and remote tissues will begin to return to more youthful function; in turn, the renewal of those remote tissues' function then further contribute to the re-establishment of youthful homeostasis in the system as a whole. As the regenerative process feeds back upon itself, accelerated with each new therapy applied and each additional form of damage repaired, the function of tissues, organs, we may hypothesize that the organism as a whole will re-emerge in unexpected ways and with unanticipated inflection points, until we stand restored to the full health, vigor, and capacity of youth.
References
1. Mayack SR, Shadrach JL, Kim FS, Wagers AJ. Systemic signals regulate ageing and rejuvenation of blood stem cell niches. Nature. 2010 Jan 28;463(7280):495-500. PubMed PMID: 20110993.
2. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001 Feb;27(2):195-200. PubMed PMID: 11175789.
3. Bartke A. Growth hormone and aging: a challenging controversy. Clin Interv Aging. 2008;3(4):659-65. Review. PubMed PMID: 19281058; PubMed Central PMCID: PMC2682398.
4. Adams MM, Elizabeth Forbes M, Constance Linville M, Riddle DR, Sonntag WE, Brunso-Bechtold JK. Stability of local brain levels of insulin-like growth factor-I in two well-characterized models of decreased plasma IGF-I. Growth Factors. 2009 Jun;27(3):181-8. PubMed PMID: 19343576.
5. Sun LY, Evans MS, Hsieh J, Panici J, Bartke A. Increased neurogenesis in dentate gyrus of long-lived Ames dwarf mice. Endocrinology. 2005 Mar;146(3):1138-44. Epub 2004 Nov 24. PubMed PMID: 15564324.
6. Sonntag WE, Lynch CD, Cefalu WT, Ingram RL, Bennett SA, Thornton PL, Khan AS. Pleiotropic effects of growth hormone and insulin-like growth factor (IGF)-1 on biological aging: inferences from moderate caloric-restricted animals. J Gerontol A Biol Sci Med Sci. 1999 Dec;54(12):B521-38. Review. PMID: 10647962
7. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005 Feb 17;433(7027):760-4. PMID: 15716955
8. Fauce SR, Jamieson BD, Chin AC, Mitsuyasu RT, Parish ST, Ng HL, Kitchen CM, Yang OO, Harley CB, Effros RB. Telomerase-based pharmacologic enhancement of antiviral function of human CD8+ T lymphocytes. J Immunol. 2008 Nov 15;181(10):7400-6. PubMed PMID: 18981163; PubMed Central PMCID: PMC2682219.
9. Ju Z, Jiang H, Jaworski M, Rathinam C, Gompf A, Klein C, Trumpp A, Rudolph KL. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat Med. 2007 Jun;13(6):742-7. Epub 2007 May 7. PubMed PMID: 17486088.
10. Passos JF, Saretzki G, von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 2007;35(22):7505-13. PMID: 17986462
11. . Sarin KY, Cheung P, Gilison D, Lee E, Tennen RI, Wang E, Artandi MK, Oro AE, Artandi SE. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature. 2005 Aug 18;436(7053):1048-52. PMID: 16107853 [PubMed - indexed for MEDLINE]
12. Flores I, Cayuela ML, Blasco MA. Effects of telomerase and telomere length on epidermal stem cell behavior. Science. 2005 Aug 19;309(5738):1253-6. Epub 2005 Jul 21. PMID: 16037417 [PubMed - indexed for MEDLINE]
13. Liu L, DiGirolamo CM, Navarro PA, Blasco MA, Keefe DL. Telomerase deficiency impairs differentiation of mesenchymal stem cells. Exp Cell Res. 2004 Mar 10;294(1):1-8. PMID: 14980495 [PubMed - indexed for MEDLINE]
14. de Grey ADNJ, Campbell FC, Dokal I, Fairbairn LJ, Graham GJ, Jahoda CAB, Porter ACG. Total deletion of in vivo telomere elongation capacity: an ambitious but possibly ultimate cure for all age-related human cancers Ann N Y Acad Sci. 2004 Jun;1019:147-70. PubMed: 15247008.
15. de Grey ADNJ. Whole-body interdiction of lengthening of telomeres: a proposal for cancer prevention. Front Biosci 2005;10:2420-2429. PubMed: 15970505.
16. de Grey AD. WILT: Necessity, feasibility, affordability. In: Fahy GM, West M, Coles LS, Harris SB (eds) The Future of Aging: Pathways to Human Life Extension. 2010; Springer, 667-684.
17. Xu X, Sonntag WE. Moderate caloric restriction prevents the age-related decline in growth hormone receptor signal transduction. J Gerontol A Biol Sci Med Sci. 1996 Mar;51(2):B167-74. PubMed PMID: 8612101.
18. Sonntag WE, Xu X, Ingram RL, D'Costa A. Moderate caloric restriction alters the subcellular distribution of somatostatin mRNA and increases growth hormone pulse amplitude in aged animals. Neuroendocrinology. 1995 May;61(5):601-8. PubMed PMID: 7617139.
19. D'Costa AP, Lenham JE, Ingram RL, Sonntag WE. Moderate caloric restriction increases type 1 IGF receptors and protein synthesis in aging rats. Mech Ageing Dev. 1993 Oct 1;71(1-2):59-71. PubMed PMID: 7508538.
Posted 03 August 2010 - 10:49 AM
The 7KC-degrading bacterium I’ve been studying, Rhodococcus jostii RHA1, has two large gene clusters that are up-regulated by 7KC, but not cholesterol. In these two gene clusters lie a number of enzymes we believe are involved in 7KC degradation, including an enzyme that could reduce the 7-keto group to a hydroxyl. What makes this interesting to us is that while 7KC is highly cytotoxic, 7α-hydroxycholesterol (7αOH) is relatively harmless. So I am now methodically going through suspected candidates, searching for reductase activity against 7KC. Currently I am looking at nine different enzymes, and am in various stages of cloning the genes into expression vectors and assaying their products for activity.
While I’ve uncovered some interesting findings, so far I haven’t found the reductase I’m looking for. This could be for several reasons, the first being that I haven’t assayed all the enzymes I need to. As I still have the majority remaining, this is a likely scenario. However, one possibility is that the normal substrate for the enzyme is not 7KC, but some downstream metabolite. If this is the case then I have several approaches I could take.
The first would be to test downstream metabolites, though this would be very time consuming as most of these compounds are not commercially available. The second approach would be to use mutagenesis techniques on the enzymes I have to see if I can alter their substrate specificity to accept 7KC. I could also systematically delete candidate genes and monitor 7KC degradation, though that could be time consuming as well. Another alternative is to obtain hamster 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD). 11β-HSD has been found to interconvert 7KC, 7βOH, and 7αOH. Unfortunately 7βOH is more cytotoxic than 7KC, so this activity isn’t beneficial, though this enzyme could potentially be engineered for altered stereoisomerism. The ideal path may be to attempt all of these approaches in parallel, but I’d like to finish subcloning and expressing the genes I have before making that decision.
Posted 06 August 2010 - 03:16 AM
Kamil Pabis is in his second year of university and has been working with the SENSFAI since 2009. He is currently studying biology at the University of Vienna.
<!--break-->  After completing his degree, Kamil plans to pursue his PhD and eventually a career in Molecular Biology or Biogerontology.
After completing his degree, Kamil plans to pursue his PhD and eventually a career in Molecular Biology or Biogerontology.
His first and current project with SENSFAI is a research paper related to vascular calcification.
You can read the text of our interview below.
I have always been interested in biology, generally all the life sciences, but on the other hand also engineering. One could say "science" in general.
The choice of my field of study basically came down to a coin toss between the closest contenders, but not because I wasn't particularly interested in any of them. To the contrary, I was highly interested in several fields. Eventually I decided to study biology among other reasons, perhaps due to gotten involved with the community at imminst.org. I'm pursuing a Bachelor's degree but plan to continue and work towards a PhD. I am a little more than halfway through.
I would like to work in the field of bio-gerontology and especially work on definitive treatments for aging, not just stop-gap solutions. However, I also want to broaden my knowledge in all the related and interesting fields e.g. medicine, molecular biology and biochemistry; engineering; life styles related to life extension and the 'human enhancement' movement.
Beyond my general interest in biology that we discussed before, the specialisation I chose came down to chance again. The number of interesting subfields in biogerontology and beyond is astounding and I could imagine working on a number of them. My first SENS project is in the field of vascular disease, so I decided to primarily, but not exclusively, focus on CVD and CV-aging.
The first thing I read about SENS were Michael Rae's exceptionally lucid and convincing contributions to this topic. I'm sure if it wasn't for his posts it would have taken me much longer to discover the merits of SENS and related ideas.
Perhaps, true to the idea that ‘any publicity is good publicity’, criticism leveled against SENS, including the Technology Review controversy, further boosted my interest and enthusiasm. Despite criticism Aubrey's approach struck me, basically an interested laymen at that time, as perfectly valid and elegant.
I research vascular (and in part general) calcification and their relation to aging and age-related tissue decline. The impact of calcification could be major and under-appreciated, but unfortunately we do not have definitive data.
This basic research lays the ground work for future projects. A relatively thorough understanding is required to distinguish the most promising therapies for actual reversal of the pathology. Eventually I plan to help facilitate and do research under a "regression first" paradigm.
I hope (and am convinced) that in the future bio-gerontologists will appreciate the idea of SENS considerably more than they do now. Therefore my involvement should have a very positive impact. And in any case it remains an eye-catching conversation starter.
I would like to work on other similar topics of course, but would also be interested in pursuing research around these topics: Tissue regeneration (limbs, scarless healing, etc), the influence of phosphate metabolism on aging and disease (this is more a general biogerontology topic), Cardiovascular Disease in general, and atherosclerosis specifically (e.g. the lysoSENS or other approaches)
Posted 15 August 2010 - 12:34 AM
Age-related accumulation of mutations in mitochondrial DNA (mtDNA) are widely suspected to play an important role in the degenerative aging process, albeit that controversy remains as to the mechanism(s) linking the two. Large deletions in mtDNA seem an especially likely culprit, due to (a) the clear, but focal, accumulation of cells homoplasmic for mtDNA deletions in postmitotic tissues; (b) their association with age-related pathology (the substantia nigra accumulates deletion-homoplasmic cells in aging and Parkinson's disease, as do skeletal muscle fiber segments in association with fiber breakage and sarcopenia, etc); and © their ability to completely abrogate oxidative phosphorylation (OXPHOS), principally because even the smallest mtDNA deletion encompasses several genes encoding mitochondrial tRNAs.
A number of credible proposals have been advanced for rejuvenation biotechnology to restore youthful mitochondrial function in such cells, reverting their abnormal metabolism and allowing them to resume participation in normal tissue function. The lead candidate approach, first proposed by SENS Foundation Chief Scientific Officer de Grey,(1) is the placement of functioning "backup copies" of the protein-coding mtDNA genes in the cell nucleus ("allotopic expression" (AE)). There has been substantial progress in this area since then,(eg (5-9)), and in recent years SENS Foundation has prioritized funding of AE research beginning with early work by Mark Hamalainen in Ian Holt's lab at Cambridge, and later in both the SENS Foundation Research Center and in the lab of Dr. Marisol Corral-Debrinski at the Institut de la Vision at Pierre and Marie Curie University, Paris(8). Active investigation of AE is soon to resume in the latter two centers.
But other potential routes to mitochondrial rejuvenation do exist and should also be developed, including the wholescale intraorganellar replacement of mtDNA using "protofection" (2) and the delivery of allotopic RNA to the organelle. The latter possibility was highligted by work targeting tRNA human cell mitochondria with the transgenic use of the transfer RNA import complex adapted from the parasitic protozoon Leishmania tropica.(3) Working with Newcastle University's Dr. Robert Lightowlers and others, UCLA's Carla Koehler and Michael Teitella have now identified and begun to characterize a mammalian-specific mitochondrial system for the import of nuclear-encoded RNA, which could well be exploited to meet this biomedical challenge.
Much is understood about the mechanisms that regulate nuclear-encoded protein import into mitochondria. By contrast, much less is known about the factors that regulate mitochondrial RNA import. Almost every organism with mitochondria imports tRNAs and aminoacyl-tRNA ... with mammalian mitochondria importing several different tRNAs both in vitro and in vivo ... RNase MRP and RNase P enzyme complexes localize and function in mammalian mitochondria and may contain RNAs that are encoded within the nucleus. ... [T]he RNA component of RNase P was imported into mammalian mitochondria and the RNase P RNA and processing activity has been copurified from mitochondria. By contrast, RNase P RNA is encoded in the mitochondrial genome in Saccharomyces cerevisiae. Recently, an additional RNase P enzyme, consisting of three protein subunits, has been purified from human mitochondria. This alternative RNase P enzyme processes single tRNA 5′ precursor sequences in vitro without an RNA component...
Previously, we localized polynucleotide phosphorylase (PNPASE), a 3′ → 5′ exoribonuclease and poly-A polymerase ["that uses phosphorolysis to degrade RNA"], in the mitochondrial intermembrane space, a location lacking resident RNAs. ... This was a surprise because we expected that PNPASE would instead localize in the RNA-abundant mitochondrial matrix. ...
Here, we show that PNPASE has a central role in augmenting the import of small RNA components required for DNA replication and RNA processing into the mitochondrial matrix. PNPASE reduction impaired mitochondrial RNA processing and polycistronic transcripts accumulated. Augmented import of RNase P, 5S rRNA, and MRP RNAs depended on PNPASE expression and PNPASE–imported RNA interactions were identified. ...
To establish a direct PNPASE role, a systematic search was used to identify PNPASE-dependent RNA import sequences. Primers were designed to generate distinct segments of the 340 nucleotide (nt) RNase P RNA full length sequence. ... Import assays were performed using full length or truncated in vitro transcribed RNase P RNAs. ... [A series of truncations and importation experiments] implicat[ed] an import signal between nt 103 and 140. The most obvious, predicted secondary structure of RNase P RNA in this region was a 20 nt stem-loop. Interestingly, a similarly-predicted stem-loop structure was also identified in MRP RNA. ... [E]ach 20 nt stem-loop sequence was fused to the 5′-terminus of the GAPDH RNA, which is not imported. Strikingly, the RNase P and MRP stem-loop structures licensed the PNPASE-dependent import of GAPDH RNA into yeast mitochondria. By contrast, a control random 20nt sequence could not mediate this import. Human mitochondrial tRNA(trp) with the RNase P RNA step-loop structure, but not tRNA(trp) itself, was imported into isolated mouse liver mitochondria ... Finally, replacement of the human RNase P RNA stem-loop sequence with the 20nt random sequence blocked augmented RNase P RNA import into yeast mitochondria in vivo, confirming the role of the stem-loop in PNPASE-regulated import.
PNPASE augmented import of RNase P RNA into yeast mitochondria is nonphysiologic. Therefore, we developed WT, PNPASE KO, WT expressing human PNPASE, and PNPASE KO expressing human PNPASE MEFs for import assays. ... . Radiolabeled RNase P RNA was not imported into mitochondria from the PNPASE KO MEFs, but was imported into mitochondria that contained mouse and/or human PNPASE. The in vitro import of RNase P, MRP, 5S rRNA, and GAPDH RNAs was also tested [to similar effect], ... whereas cytosolic GAPDH RNA was not imported. As expected, more than half of the imported MRP RNA was processed into the mature ∼130 nt form. By contrast, mitochondrial RNA import was severely compromised in HepKO liver mitochondria. Combined, these results strongly support PNPASE as the first RNA import factor that mediates the translocation of specific RNAs into the mammalian mitochondrial matrix.
These results strongly implicate the structural specificity of mitochondrial RNA import and the direct involvement of PNPASE in this process. ... PNPASE RNA processing and import activities were separable and a mitochondrial RNA targeting signal was isolated that enabled RNA import in a PNPASE-dependent manner. Combined, these data strongly support an unanticipated role for PNPASE in mediating the translocation of RNAs into mitochondria. (4)
This study makes a significant contribution to our understanding of the mechanisms of mitochondrial RNA import of nuclear-encoded RNA in mammals. And although it is just the first step along the way, in principle this discovery also suggests a way to bypass the paralysis of mitochondrial protein synthesis secondary to large mtDNA deletions. Dr. Adhya's work with the Leishmania transfer RNA import complex (3) for which reason he was given a prominent place at the 3rd SENS Scientific Conference, had already suggested the possibility of delivering the full spectrum of mitochondrially-encoded RNAs into the mitochondrion from "backup copies" of the underlying genes housed in the nucleus. Pursuit of the same strategy through the exploitation of the native mammalian RNA import machinery would be expected to achieve the same outcome in a more elegant and facile manner. In principle, all that might be required is the fusion of the RNase P stem-loop structure on to the allotopically-expressed genes for the full spectrum of mitochondrially-encoded RNA species. Such constructs would be designed to allow for their transcription without cytosolic translation by noninclusion of a 5′UTR. Conceivably, the efficiency of mitochondrial delivery could be enhanced if a segment of the 5′UTR exploited by Dr. Corral-Debrinski's group could be identified that would not allow for translation, but would still preserve its ability to facilitate translocation to the mitochondrial surface.
And of course, in principle there is no exclusivity between AE of protein and AE of mRNA: provided that at minimum the mitochondrial tRNAs can be allotopically expressed and imported, regenerative engineers could deliver some fully-translated AE proteins and some mRNA precursors, depending on the facility and efficiency of either approach for a given protein, and on the burden on the relevant import machinery.
This is a preliminary sketch of a possible future rejuvenation biotechnology, and one that the investigators evidently had not contemplated. Their own attention at this time is focused on the more immediate basic science questions of "what other pathway components are involved and what the RNA sequence or structure rules tell us about how PNPASE may decipher between processing and import."(4) But it opens up the potential of a new way to achieve one of the goals necessary for the function and structure of tissues, organs, cells, and (yes) organelles to be restored to youthful norms.
References
1. de Grey AD. A mechanism proposed to explain the rise in oxidative stress during aging. J Anti-Aging Med 1998;1(1):53-66.
2.Khan SM, Bennett JP Jr. Development of mitochondrial gene replacement therapy. J Bioenerg Biomembr. 2004 Aug;36(4):387-93. Review. PubMed PMID: 15377877.
3. Mahata B, Mukherjee S, Mishra S, Bandyopadhyay A, Adhya S. Functional delivery of a cytosolic tRNA into mutant mitochondria of human cells. Science. 2006 Oct 20;314(5798):471-4. PubMed PMID: 17053148.
4. Wang G, Chen HW, Oktay Y, Zhang J, Allen EL, Smith GM, Fan KC, Hong JS, French SW, McCaffery JM, Lightowlers RN, Morse HC 3rd, Koehler CM, Teitell MA. PNPASE Regulates RNA Import into Mitochondria. Cell. 2010 Aug 6;142(3):456-467. PubMed PMID: 20691904.
5. Zullo SJ, Parks WT, Chloupkova M, Wei B, Weiner H, Fenton WA, Eisenstadt JM, Merril CR. Stable transformation of CHO Cells and human NARP cybrids confers oligomycin resistance (oli®) following transfer of a mitochondrial DNA-encoded oli® ATPase6 gene to the nuclear genome: a model system for mtDNA gene therapy. Rejuvenation Res. 2005 Spring;8(1):18-28. PubMed PMID: 15798371.
6. Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, Schon EA. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002 Apr;30(4):394-9. Epub 2002 Feb 25. PubMed PMID: 11925565.
7. Guy J, Qi X, Pallotti F, Schon EA, Manfredi G, Carelli V, Martinuzzi A, Hauswirth WW, Lewin AS. Rescue of a mitochondrial deficiency causing Leber Hereditary Optic Neuropathy. Ann Neurol. 2002 Nov;52(5):534-42. PubMed PMID: 12402249.
8. Bonnet C, Augustin S, Ellouze S, Bénit P, Bouaita A, Rustin P, Sahel JA, Corral-Debrinski M. The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim Biophys Acta. 2008 Oct;1783(10):1707-17. Epub 2008 May 6. PubMed PMID: 18513491.
9. Supekova L, Supek F, Greer JE, Schultz PG. A single mutation in the first transmembrane domain of yeast COX2 enables its allotopic expression. Proc Natl Acad Sci U S A. 2010 Mar 16;107(11):5047-52. Epub 2010 Mar 1. PubMed PMID: 20194738.
Posted 23 August 2010 - 09:12 PM
A comprehensive suite of rejuvenation biotechnologies must include the removal of extracellular aggregates from aging cells and tissues. The most clinically-advanced such biotechnology is immunotherapy against aggregated beta-amyloid protein (Aβ), a characteristic neuropathological lesion that accumulates in the brain in Alzheimer's disease (AD) patients and as part of "normal" brain aging.(1)
The promise of active and passive Aβ-targeting vaccines is high, but experimental and clinical testing of such therapies have revealed some of their limitations. Immunotherapeutics currently in clinical development rely in different ways on the mobilization of the patient's immune processes. Active Aβ immunotherapy (such as AN1792) involvesaggregates of Aβ itself (along with adjuvants), mobilizing an active Aβ-targeting immunoglobulin G (IgG) antibody response. Therapeutic efficacy thus depends on the patient's immune response to vaccination, which notoriously declines with aging; indeed, in the phase I trial of AN1792, serum anti-Aβ antibody titers were low, exceeding the secondarily-defined threshold of 1:1,000 in less than a quarter of AD patients. This low response was enhanced in the later Phase IIa trial by reformulation of the adjuvant, which increased the immunological response rate to nearly 60%, but generated a shift from a predominantly Th2 response to an inflammatory Th1 T-cell response. Unfortunately, this aggressive T-cell response led to the emergence of meningoencephalitis in ~6% of patients, leading to the early cessation of the trial.(1) Second-generation active vaccines are in development that are designed to minimize these risks, but may to varying degrees necessitate a difficult balance of pro- and noninflammatory mechanisms. Moreover, should these or other side effects occur as a result of active vaccination, host immune response will persist for some time until anti-Aβ antibody titers decay, potentially requiring the extended administration of immunosuppressive therapy to avert major adverse events.
Passive Aβ-targeting vaccines (bapineuzumab is furthest down the clinical pipeline) administer such IgGs generated ex vivo directly to patients, eliminating the risks associated with activation of host active immune response, and allowing for immediate cessation of therapy should adverse events occur. But in clinical trials, passive vaccination with bapineuzumab has exhibited side-effects of its own, causing vascular microhaemorrhages in a minority of apolipoprotein E ε4 carriers, thought to be caused by IgG-bound Aβ monomers or small oligomers being deposited in the cerebral vasculature following mobilization from stable plaques;(1) such effects might be exacerbated by the relative stability of the IgG-Aβ immune complexes as they bind to BBB receptors, and the activation of inflammatory mediators.(4) Parallel effects have been observed following passive vaccination in transgenic mouse models of AD.
Moreover, monoclonal Aβ-targeting antibodies are by their nature quite expensive, and are likely to require large (stoichiometric) quantities of IgG antibodies to be administered on a regular and frequent schedule to maintain therapeutic levels of the antibodies in the patient. These features might make the cost of ongoing therapy prohibitive, especially for an extended schedule of preventive therapy in the large number of"normally"-aging individuals with significant Aβ deposition but without existing cognitive impairment who are in principle the best candidates for such therapy.(3) Anticipated frequencies of dose administration could also be problematic: an early Phase I trial of bapineuzumab, for instance, estimated on the basis of pharmacokinetic data that a 13-week dosing interval would be necessary to secure ongoing therapeutic benefit.(4) In addition to requiring large financial outlays for both the immunotherapy agent itself and for the health professionals who would administer them, frequent dosing would significantly inconvenience to patients, likely leading to noncompliance in asymptomatic patients.
An ideal Aβ immunotherapy would thus not depend on the patient's immune system for effectiveness or safety, but would have an "intrinsic" mechanism of action, and would have a high ratio of aggregate clearance to antibody dose counts and frequency of administration.
Dr. Sudhir Paul and his group at the Chemical Immunology Research Center at the University of Texas-Houston Medical School have made significant progress with a promising novel approach to Aβ immunotherapy that promises to deliver on all of these fronts. They have identified, purified, and characterized endogenous catalytic antibodies that with direct hydrolytic activity against these pathological aggregates, and have isolated rare chain fragments that are even more potent; they are advancing there research progressively toward developing these antibody fragments into a therapy for AD and brain aging.
Catalytic Immunotherapy Against Beta-Amyloid
As Dr. Paul's group report,
Michael Sierks at the University of Arizona observed that two recombinant antibody light-chain subunits with [vasoactive intestinal peptide]-hydrolyzing activity also displayed cross-reactive Aβ-hydrolyzing activity. This prompted us to screen monoclonal and polyclonal preparations of intact antibodies for this activity. ... Naturally occurring immunoglobulinM (IgM) class antibodies that hydrolyze Aβ and inhibit Aβ aggregation were identified. ... Most IgM preparations from nondemented elderly humans displayed detectableAβ40 hydrolytic activity. ... The production of these antibodies increases as a function of age, ostensibly reflecting an attempt by the immune system to protect against the deleterious effect of Aβ accumulation in old age. [Similarly, "Twenty two of the 25 IgM preparations from undemented elderly humans studied displayed detectable (125)I-Aβ40 hydrolytic activity varying over a 118-fold range. [Fig. 1(A) below]... IgMs from [an age-matched] AD group displayed superior hydrolytic activity [Fig. 1(B) below]"(5)] ...
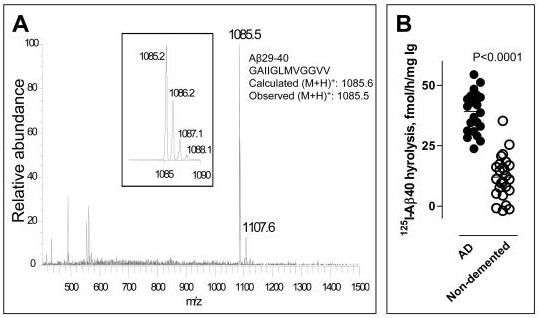
Figure 1: Variation in Aβ-catalytic activity of IgM isolated from nondemented elderly (1(A)) and in that from AD patients (n=23, Fig 1(B), left). vs. elderly, nondemented controls (n=25, right). (From (4))
As previously discussed, purified, pooled human immunoglobulins for intravenous delivery (IVIG) have already shown evidence of efficacy as an anti-Aβ immunotherapy. It would be reasonable to assume that IVIG would include Aβ-hydrolyzing IgM clones. However, Paul's group has found no evidence of such activity:
IgG purified from the plasma of old humans by an affinity fractionation procedure involving acid treatment also hydrolyzed Aβ detectably, albeit at levels smaller than the IgM fractions from the same humans. [Yet] No Aβ hydrolysis was detected by [either of 2 approved] IVIG preparations ... [Nonspecific small model] peptides were also cleaved poorly by IVIG preparations compared with IgG purified by the acid-affinity purification procedure. Evidently, the IgG catalytic activity does not survive the purification procedures used to prepare IVIG. Recent studies on a recombinant catalytic antibody fragment also suggest the sensitivity of the catalytic site to conformational perturbations ...(5)
We searched for Aβ -hydrolyzing recombinant human immunoglobulin variable (IgV) domains in a library composed of ~10[to the power of]7 clones from humans with systemic lupus erythematosus, an autoimmune disease associated with enhanced production of catalytic antibodies ... A minority of clones possess unusual IgV structures generated by DNA manipulation errors that inevitably accompany repeated nucleic acid replication and cloning cycles over the course of library construction. By random screening and covalent phage-IgV selection using an Aβ40 analog ... we isolated rare IgV clones capable of hydrolyzing Aβ40 rapidly. The rate of Aβ hydrolysis by the IgVs was 3-4 orders of magnitude greater than the naturally occurring IgMs … For example, from its turnover number determined at excess Aβ concentration, a single [candidate catalytic antibody fragment] molecule will degrade 4320 Aβ molecules in 3 days. The rate of catalysis is comparable to that of neprilysin, an enzyme that has received attention as a potential Aβ-clearing drug. In unpublished studies, we observed that minimizing conformational perturbations of the IgVs during purification improves the catalysis rate by another order of magnitude....(2)
The IgVs are also unique by virtue of their specificity for Aβ. ... The IgVs did not hydrolyze irrelevant proteins and model protease substrates customarily employed to monitor catalyst specificity … suggesting the feasibility of specific Aβ clearance with little or no damage to other proteins. ... Neprilysin and other Aβ-hydrolyzing enzymes, in contrast, hydrolyze irrelevant polypeptides. (2)
The fact that these IgV clones hydrolyze, rather than sequester, Aβ offers numerous theoretical advantages over immunotherapies currently in clinical trials. First, it could dramatically reduce the amount of antibody required for Aβ clearance relative to passive immunization, because a small number of high-activity catalytic enzyme fragments could hydrolyze a large number of Aβ molecules, likely reducing the cost of each round of therapy. Secondly, because the enzyme has autonomous hydrolytic activity, rather than relying on other immunologic response, it should not be limited by the immune status of recipients, nor cause inflammatory or other immunologic side-effects. Moreover, the effectiveness of a catalytic IgV immunotherapy should powe much less risk of vasogenic edema as a consequence of sequestration and efflux of IgG-bound Aβ: the aggregates would be permanently hydrolyzed, rather than sequestered into stable immune complexes and then transported through the cerebral vasculature and out through the BBB, and then left to circulate systemically before ultimate nonspecific degradation in the circulation or disposal by excretion.(2,5) (Figure 2) And, finally, the high specificity of the IgV clones for Aβ further implies an absence of off-target adverse effects.
"Catalytic IgM preparations from healthy humans inhibited Aβ aggregation, dissolved preformed Aβ aggregates, and inhibited the toxic effect of Aβ oligomers on cultured neuronal cells."(5) And there is preliminary evidence of efficacy in a mouse model of AD, based on isolated human Aβ-cleaving IgM rather than the more therapeutically optimized IgV clones:
In a preliminary study, a preparation of catalytic human IgM from pooled human serum was administered intravenously on day 0 and day 8 to 6 month old APP-Tg mice that overexpress human Aβ (APPSwe/PS1ΔE9 mouse strain). A sustained increase of intact Aβ concentrations in peripheral blood determined was evident [Fig 3 below]. As the injected human IgM did not bind Aβ detectably, the evident increase of pepripheral Aβ is not due to peptide stabilization by formation of immune complexes. This suggests the feasibility of depleting brain Aβ as a consequence of peripheral IgM catalyzed Aβ hydrolysis."(5)
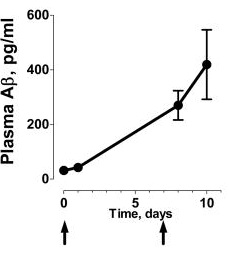
Figure 3 Increase in peripheral Aβ in TG AD model mice following injection of human Aβ-targeting IgM. From (5)
Since the catalytic IgM pools did not form stable immune complexes, the conventional mechanisms for antibody-mediated efflux of Aβ from the CNS could not apply. Dr. Paul's group proposed mechanism of this peripheral mobilization is thus distinct from that apparaently in place with sequestering IgG-based immunotherapies (active and passive), leading to anticipated therapeutic and safety advantages, as illustrated in Figure 4:
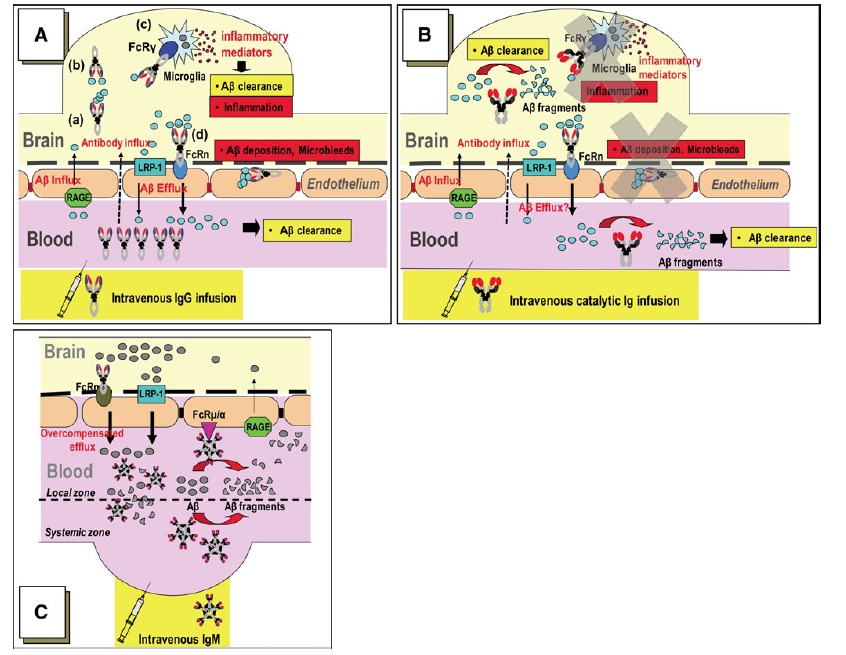
Figure 4 comparison of mechanisms of BBB Aβ efflux by peripheral immunotherapies. (A): Aβ sequestered by IgG are taken up by microglial Fcγ receptors for digestion, and IgG-bound Aβ may be taken up by neonatal Fc receptors (FcRn) at the BBB, leading to BBB transcytosis and peripheral efflux. The former is likely responsible for the inflammation associated with active vaccination with AN1792; the latter appears to contribute to vasogenic edema following deposition of stable complexes in the cerebral vasculature. (B): catalytic IgM or IgM-derived IgV clones should abrogate these potentially deleterious effects. Instead, © on the basis of the observed increase in peripheral Aβ in Alzheimer's model mice administered human Aβ-targeting IgM, Dr. Paul's group hypothesizes that as Aβ hydrolysis in the periphery increases the trans-BBB Aβ concentration gradient, IgM binds to BBB Fc receptors for IgM (Fcμ/α) and promotes direct Aβ transcytosis by FcRn. (Figure from (6)
Aβ-targeting IgV clones remain in the early stages of development, placing it in a crowded field of other recently-developed anti-Aβ immunotherapies as candidates that appear to be approaching the exit spout justifiably draw the greatest amount of attention. But its novel, molecule-"autonomous" mechanism of action is more in line with the core principles of regenerative engineering, and in principle offers a high promise of therapeutic efficacy, attractiveness for preventive use in nondemented older adults, and little risk of adverse reactions known or anticipated in others far further developed. Their approach thus merits careful attention -- and even cautious excitement -- in its own right.
References
1. Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
2. Paul S, Planque S, Nishiyama Y. Beneficial catalytic immunity to Abeta peptide. Rejuvenation Res. 2010 Apr-Jun;13(2-3):179-87. Review. PubMed PMID: 20370602.
3. Cribbs DH. Aß DNA vaccination for Alzheimer's disease: focus on disease prevention. CNS Neurol Disord Drug Targets. 2010 Apr;9(2):207-16. PubMed PMID: 20205639.
4. Black RS, Sperling RA, Safirstein B, Motter RN, Pallay A, Nichols A, Grundman M. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 2010 Apr-Jun;24(2):198-203. PubMed PMID: 20505438.
5. Taguchi H, Planque S, Nishiyama Y, Szabo P, Weksler ME, Friedland RP, Paul S. Catalytic antibodies to amyloid beta peptide in defense against Alzheimer disease. Autoimmun Rev. 2008 May;7(5):391-7. Epub 2008 Apr 9. Review. PubMed PMID: 18486927; PubMed Central PMCID: PMC2430036.
6. Paul S, Planque S, Nishiyama Y. Immunological origin and functional properties of catalytic autoantibodies to amyloid beta peptide. J Clin Immunol. 2010 May;30 Suppl 1:S43-9. PubMed PMID: 20454852.
Posted 28 August 2010 - 10:05 AM
On behalf of SENS Foundation, I'm pleased to announce the launch of a competitive research prize for "Breaking Advanced Glycation Endproduct Glucosepane, a Protein Cross-link" through Innocentive, a global leader in open innovation.
As we work to accelerate the development of a comprehensive suite of rejuvenation biotechnologies, a key milestone will be a therapy that cleaves Advanced Glycation Endproduct (AGE) crosslinks. As we live and process carbohydrate and fat, side-reactions in our bodies cause these fuels to react with the proteins in our cells, tissues, and organs, slowly stiffening them so that they lose the elastic responsiveness that lets them carry out their essential, youthful function. The accumulation of these crosslinks in our major arteries contributes to a steady rise in blood pressure as we age, leading to strokes, loss of kidney function, and abnormal enlargement and weakening of the heart muscle; they "AGE" many other organs in more subtle ways, and are a key contributor to the many terrible complications of diabetes. Severing these molecular handcuffs will be critical to the rejuvenation of aging tissues.
As a biomedical charity, SENS Foundation's usual method for tackling research problems is to provide direct grants for expert researchers to do critical-path work in rejuvenation science. So we have long had an open Request for Proposals (RFP) for qualified researchers to tackle this problem, addressing specifically the most important of those crosslinks -- a stubborn AGE called glucosepane. To date, we've had no takers.
That's why we've launched this $20,000 Theoretical Research prize -- not to demonstrate the breakage of glucosepane in the lab, but to give us a clear enough roadmap for the project that we can attract the further financial and scientific resources needed for a full-scale research and development project.
Now, with the critics exhausted, we've put up another $20,000 for a more constructive goal. SENS Foundation is reaching out to InnoCentive's network of more than 200,000 Solvers: in the next 60 days, give us a detailed working plan to develop a drug to give aging arteries their youthful spring, and averting age-related disease and pathology.
"Of all protein crosslinks, glucosepane makes the single largest contribution to the stiffening of aging tissues in humans, so finding a way to break it down is our Foundation's top priority within that subset of targets," said Aubrey de Grey, Ph.D., Chief Science Officer of SENS Foundation. "We believe there are several radical possibilities to solving this problem -- things we haven't even thought of -- and will keep an open mind to solutions we receive. If one of Innocentive's network of 200,000 Solvers can help us find a design for a successful program, it would be an important keystone of a critical goal: reversing stiffening, and restoring youthful elasticity and vigor to tissues. We thank Innocentive for working with us on this Prize, and for bringing their network to bear on the challenge."
"There are astounding implications to the possibility of reversing aging and living a longer, healthy life," said Dwayne Spradlin, CEO of InnoCentive. "SENS has identified seven categories that cause us to age. While this Challenge addresses only one type of crosslink in one category, if our Solver community can solve this Challenge, it could help SENS figure out how to attack the other types of crosslinks."
Mike Kope
Chief Executive Officer, SENS Foundation
SENS Foundation is a nonprofit medical research organization which works to develop, promote and ensure widespread access to rejuvenation biotechnologies. Our efforts combine direct research efforts with education, affiliation and outreach programs. We support key research through our internal Research Center and through centers of excellence at a variety of affiliated universities and research organizations. Our vision is a world in which all people have the opportunity to live their lives free from the diseases and disabilities of aging. http://www.sens.org/
Since 2001, InnoCentive has helped corporate, government, and non-profit organizations to better innovate through crowdsourcing, strategic consulting services and internal Software-as-a-Service offerings.The company built the first global Web community for open innovation where organizations or "Seekers" submit complex problems or "Challenges" for resolution to a "Solver" community of more than 200,000 engineers, scientists, inventors, business professionals, and research organizations in more than 200 countries. Prizes for winning solutions are financial awards up to US $1,000,000. Committed to unleashing diverse thinking, InnoCentive continues to introduce new products and services exemplifying a new corporate model where return to investors and individual passion go hand in hand with solving mankind's most pressing problems. http://www2.innocentive.com/
References
Sjöberg JS, Bulterijs S. Characteristics, formation, and pathophysiology of glucosepane: a major protein cross-link. Rejuvenation Res. 2009 Apr;12(2):137-48. Review. PubMed PMID: 19415980.
de Grey AD. Foreseeable pharmaceutical repair of age-related extracellular damage. Curr Drug Targets. 2006 Nov;7(11):1469-77. Review. PubMed PMID: 17100587.
Posted 04 September 2010 - 01:06 AM
In addition to its widely-anticipated potential to provide highly-effective therapies for genetic disorders, somatic gene therapy is an essential enabling technology for the repair or obviation of several of the cellular and molecular lesions driving age-related disease and dysfunction (notably the accumulations of mutations in mitochondrial and nuclear DNA). One of the most promising routes to somatic gene therapy is zinc finger nucleases (ZFNs), engineered DNA-binding proteins consisting of a FokI restriction enzyme catalytic core bookmarked into a dimer of zinc finger array DNA binding domains. The choice of zinc finger domains allows the engineer to target twinned 9-18 base-pair sequences in the recipient genome, separated from each other by a (typically) 5-7 base pair spacer. Upon binding, the restriction enzyme dimerizes, creating a double-strand break at the spacer locus; the engineer then takes advantage of the native DNA repair machinery to insert an user-supplied DNA repair template through Non-Homologous End Joining (NHEJ).

Image © Sigma-Aldrich Zinc Finger Nuclease Learning Center
One significant barrier to the engineering of specific ZFNs has been the difficulty in engineering new target sites within the host genome. To facilitate the targeting of new sites, a collaboration between Drs. Dan Voytas of the University of Minnesota Center for Genome Engineering and Adam J. Bogdanove of the Department of Plant Pathology at Iowa State University, engineered a new system for the targeted introduction of DSBs in host genomes by inserting the ZFN endonuclease into the homing system used by transcription activator-like effectors (TALEs), a family of plant pathogen virulence factors. TALEs bind to host-cell DNA and transcriptionally activate host genes that enhance pathogen virulence (or, in some cases, host defense). As Dr. Bogdanove had discovered, the TALE targeting system consists of central repetitive regions of varying repeat number and sequence but with specificity determined by two adjacent "repeat variable diresidues" (RVDs), primarily at repeat positions 12 and 13, with direct correspondence to host genome target site nucleosides.(1) By exploiting the simple, predictable target specificities of engineered TALEs, they looked to target the ZFN restriction enzyme to novel sites for facile genome editing.
The authors inserted the ZFN endonuclease into either the AvrBs3 TALE from the pepper-plant pathogen Xanthomonas campestris pv. vesicatoria or the PthXo1 TALE from the rice pathogen X. oryzae pv. oryzae at a restriction fragment unencumbered by the transcriptional activation domain. They then performed preliminary experiments with the newly-christened "TALE Nuclease (TALEN)" system with an initial, exploratory choice of a 15 base pair spacer size, which fortuitously turned out to be a shared optimum spacer length between the two TALENs (in addition to other, pathogen-specific optima) and yielded robust gene-targeting activity in a yeast lacZ reporter assay.
The researchers then went on to demonstrate the ability of engineered TALENs to target novel host target genes, using genes previously used to demonstrate site-directed mutagenesis by ZFNs: ADH1 in Arabidopsis, and more intriguingly the zebrafish Gridlock gene. They selected RVDs suitable to target coding regions within these genes with sequences consistent with the TALE targeting system parameters, removed a repeat domain from the native TALE genome, replaced it with these RVDs in proper order for gene targeting, and finally inserted the ZFN restriction enzyme into the TALE BamHI restriction fragment.
The resulting TALEN was tested against the target genes in the yeast lacZ assay, using the same DNA binding sites on either side of the gene. While artificial, this assay demonstrated the ability of the TALENs to specifically target these selected genes for site-directed mutagenesis, with robust activity in 2 such TALENs and more modest activity in a third.
The authors note how much work remains to be done to make practical use of the TALEN system:
the failure of some custom TALENs suggests that yet unknown rules govern the assembly of functional repeat domains. For example, repeat composition may influence protein stability, or interactions among repeat domains may affect DNA binding activity as has been observed for finger-finger interactions in zinc finger arrays . Alternatively, the spacer lengths we used may have prevented dimerization of [the ZFN endonuclease], as appeared to be the case for some spacers with AvrBs3. Clearly, it will be important to gain a better understanding of the relationship of spacer length to function for TALENs with different repeat domains. Ascertaining the minimal DNA binding domain might help accomplish this; however, we believe the repeats alone are not sufficient for adequate DNA binding, as TALENs constructed with just the repeat domain did not function in the yeast assay (data not shown). In the short-term, we will test whether custom TALENs can be created that recognize and cleave endogenous chromosomal targets, and we will evaluate the efficiency with which custom TALENs create genome modifications by non-homologous end-joining and homologous recombination. Such experiments will be key to assessing the full utility of these reagents for eukaryotic genome engineering.
The authors are duly cautious too about the ultimate use of their new system, not suggesting the possibility of its eventual use in mammalian systems, let alone for therapeutic use. But should they be able to demonstrate the targeting of native genes in situ, utilizing the binding sites actually present in the host genome, this paper will be remembered as a landmark along the way to precise repair of defective genes in inherited disease, and of renovation of the genome for the rejuvenation of aging tissues.
References
1: Bogdanove AJ, Schornack S, Lahaye T. TAL effectors: finding plant genes for disease and defense. Curr Opin Plant Biol. 2010 Aug;13(4):394-401. Epub 2010 Jun 1. PubMed PMID: 20570209.
2: Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. TAL Effector Nucleases Create Targeted DNA Double-strand Breaks. Genetics. 2010 Jul 26. [Epub ahead of print] PubMed PMID: 20660643.
Posted 20 September 2010 - 12:32 AM
A comprehensive suite of rejuvenation biotechnologies must include therapies that remove extracellular aggregates from aging cells and tissues. Of the biotechnologies in this class, immunotherapy against beta-amyloid (Aβ) is the furthest advanced along the clinical development pipeline.(1) The reasons for the pace of progress toward such therapies include the devastating impact of Alzheimer's disease (to whose etiology and pathogenesis Aβ is generally accepted to be a major contributor) on victims and their families, but also its relatively recent designation as a distinct "disease," and the widespread recognition of the rapidly-rising prevalence of Alzheimer's disease due to the aging of global populations, since the biological aging process is by far the most important important driver of its clinical course.
The Progressive Unmasking of A Hidden Disease
At the other extreme is the very slow rate of progress to date toward the development of therapies against the aggregates responsible for age-related cardiac amyloidoses: senile systemic amyloidosis (SSA), caused by aggregated wild-type transthyretin (TTR), and isolated atrial amyloidosis (IAA), caused by aggregated atrial natriureptide (ANP).(2,3) The prevalence of these cardiac amyloidoses is very low in middle age and early seniority (the youngest patient reported to have clinically-significant SSA was 57 years of age), but rises rapidly with each decade beginning at age 60, reaching 25% of persons ≥ 80 years.(2,3) Thus, the first cases of SSA are now beginning to appear in the postwar "Baby Boom" generation in the United States and elsewhere, and the prevalence and severity of the disease will rise further as they age; soon, the same will begin to occur elsewhere in the industrialized world, and soon thereafter in developing countries, the world undergoes a projected long period of global demographic aging.(10)
Cardiac amyloidosis infiltrates and disturbs the structural integrity of the heart muscle, as well as interfering with local cellular receptors and exerting direct cytotoxicity.(2) Even preclinical SSA exacerbates the morbidity of these diseases, for which it is often comorbid. And the disease is difficult to recognize: although the common clinical feature of age-related cardiac amyloidoses is a predominantly right-sided heart failure, the principle clinical signs and symptoms of SSA are nonspecific, and are commonly misdiagnosed as garden-variety congestive heart failure, cardiomyopathy, valvular heart disease, arrhythmia, or even coronary heart disease.(2)
Unfortunately, while scintigraphy with 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid can be used to distinguish TTR-based cardiac amyloidosis from light-chain amyloidosis, the most reliable method of diagnosis today is through an invasive cardiac biopsy, followed by either staining with Congo red or binding with Thioflavin-T. Neither method suitable for routine clinical use, and clinicians are only likely to resort to either in the unlikely event that the clinician suspects cardiac amyloidosis rather than some more common cause of heart dysfunction. Misdiagnosis of the cause of the degeneration of patient hearts is therefore common, and leads to inappropriate medical therapy of what are actually only minor contributors to the underlying disease, resulting in treatment that is inefective at best and which, at worst, contributes to iatrogenic illness.
Even if SSA is correctly diagnosed, there is little that clinicians can do for patients today: there are currently no effective, disease-specific therapies available.
The full, ominous potential of SSA as a cause of age-related disability and death has been highlighted by the unique work of the Supercentenarian Research Foundation (SRF), who study the biology of human aging through extensive characterization of the health, disease, and causes of deaths of persons who have survived to ages ≥110 y. A key part of SRF's scientific partnership with these Olympians of aging is the consensual autopsy of supercentenarians when they do ultimately die -- critical work, because autopsies are only rarely performed in persons past the seventh decade of life, because of the ageist assumption that "people that old" "just die" of "natural causes" whose specific diagnosis is of low priority.
Instead, as early organizers Dr. Stephen Coles and Elliot Bergman realized in the early years of this century, the opposite is true: when a person has survived for so long without being taken from their loved ones by the most common diseases of aging, there is an enormous potential for us to learn about unguessed-at causes of age-related disability and death that will become core targets of future rejuvenation biotechnologies, as the prominent causes of disease and disability in their "normally"-aging cohorts today are successively defeated.
While only a limited number of such detailed pathological studies have yet been performed on centenarians, and just 11 with supercentenarians, results to date have been highly suggestive even as they are preliminary: "The most significant factor in the death of seven of these individuals was TTR senile systemic amyloidosis [my emphasis]. Supercentenarians have only a 50% chance of living for one year. Successful treatment of supercentenarians for TTR amyloidosis would improve their health, their quality of life, and their lifespan."(4)
Thus, there is already a largely-unrecognized burden of SSA-related morbidity and mortality in the population today, both as a principle cause of death and disease and as a contributor to the total dysfunction of the aging cardiovascular system. And the situation will worsen globally over the course of the next 20 to 50 years, beginning in the industrialized world and the sequentially emerging in China, India, Mexico, the Middle East and beyond; ironically, this will be particularly so to the degree that other diseases are better-managed by progress in conventional medicine, or cured by rejuvenation biotechnology.
These features make the removal of wild-type TTR aggregates not only a concern of SRF as a research organization, but a key priority for SENS Foundation as a biomedical charity dedicated to accelerating the development of rejuvenation biotechnology. Accordingly, SENS Foundation has had an open request for proposals (RFP) for research toward the development of TTR-clearing therapies for several years now, and has approached several prominent researchers in the genetic amyloidosis field to submit applications for funding. Until recently, however, the Foundation had not received any strong proposals. But thanks to the efforts of SRF's Stan Primmer, such a proposal has recently come together and the Foundation has been given the opportunity to fund it -- and I am pleased to have the privilege of announcing that the proposal has been vetted, approved, and funded, and preliminary work will soon be under way.
September 2009: Leaks in the Therapeutic Pipeline
Because of his involvement with SRF, Primmer had long been very conscious of the clinical burden of SSA, and had surveyed the landscape of possible therapeutic approaches. But by the fall of 2009, as a result of his discussions with the present author and SENS Foundation Chief Science Officer Dr. Aubrey de Grey. he had become more conscious of the limitations of these approaches.(4)
First, very few of the therapies currently in development even for aggregates of mutant TTR would directly remove amyloid fibrils from the patient's tissues. But because TTR amyloids accumulate progressively with age in the heart and other organs, therapies that merely retard or even prevent the formation or deposition of new aggregates do nothing to restore the healthy, normal functionality of the organ in persons whose cardiac function is already substantially impaired, or approaching the threshold of clinical significance. It is this feature of much of current, risk-factor-based medical research that limits its effectiveness against age-related disease, and which therefore needs to be replaced by an "engineering" heuristic focused on the actual removal, repair, replacement, or rendering harmless of the cellular and molecular damage of aging, in order to rejuvenate tissues instead of decelerating their age-related decline.
But there are also significant restrictions on the likely efficacy even of those few TTR-targeting therapies that were then in early-stage development that would (conceptually) remove existing TTR aggregates. Notably, all of them depended mechanistically upon the mobilization of the patient's immune processes.(5-8) This implies that their ability to remove aggregates in would be significantly limited by the progressive decline in immune function that occurs as a result of the biological aging process, precisely in those persons most in need of therapy. This problem has in fact emerged with the Aβ-targeting immunotherapies in the most advanced stages of development, which are active and passive Aβ-targeting immunoglobulin G (IgG) vaccines. In the phase I trial of the active anti-Aβ vaccine AN1792, for example, serum anti-Aβ antibody titers were low, exceeding the secondarily-defined threshold of 1:1,000 in less than a quarter of AD patients, but it was only in such immunological responders that efficacy was demonstrated.(1) This low response was enhanced in the later Phase IIa trial by reformulation of the adjuvant, which increased the immunological response rate to nearly 60%, but generated a shift from a predominantly Th2 response to an inflammatory Th1 T-cell response. Unfortunately, this aggressive T-cell response led to the emergence of meningoencephalitis in ~6% of patients, leading to the early cessation of the trial.(1) In Phase I testing, passive vaccination with bapineuzumab has caused vascular microhaemorrhages in a minority of apolipoprotein E ε4 carriers, thought to be caused by deposition of IgG-bound Aβ in the cerebral vasculature.(1)
A similar constraint was in evidence in preclinical testing of a TTR-targeting antibody (in an animal model of a cross-reacting amyloidosis) when it included aged (>18 months) and immunodeficient (SCID) mice as a comparison to the standard cohort of young animals.(5) In young animals, injected antibodies rapidly bound to amyloid tumors, leading to rapid fibrillogenic tumor dissolution. Detailed study of this process revealed extensive infiltration of aggregates by neutrophils shortly after antibody opsonization, which they hypothesized to result from attraction and activation of neutrophils through their Fcγ receptors and consequent release of proteolytic enzymes and/or reactive oxygen species. But in aged and immunodeficient mice, amyloid dissolution was still incomplete at the end of the 3 mo study. (5)
An ideal therapy would therefore not depend on the patient's immune status for effectiveness or safety, but would be in itself sufficient for therapeutic benefit.
As an additional concern, all TTR-targeting immunotherapies currently in preclinical testing are relatively nonspecific, either binding to off-target molecules, or only binding to the mutant TTR protein and its aggregates to the neglect of the aggregates of wild-type protein underlying SSA.
Thanks to a dinner meeting that SRF's Primmer enjoyed with a key researcher during their attendance at the Fourth SENS Scientific Conference (SENS4), the construction of a new, robust pipeline is now underway.
Project: Develop Catalytic Antibodies Against Wild-Type TTR
Primmer had been seated at dinner at a table with Dr. Sudhir Paul, who was presenting the work that he and his group at the Chemical Immunology Research Center at the University of Texas-Houston Medical School at the meeting. As we reviewed in a previous posting, Paul's group have made significant progress with a promising novel approach to Aβ immunotherapy: catalytic Aβ-targeting antibody fragments that cleave, rather than sequester, these pathological aggregates.
In the course of conversation, Primmer mentioned the SRF's focus on the biomedical challenge of developing new diagnostics and therapies against TTR, for the health of both the extreme survivors with whom he works and the general aging population. Primmer had missed Dr. Paul's presentation, and Paul did not interject at the time to outline his work with Aβ. But Paul later realized the potential to extend his own methods, developed while researching his Aβ-targeting antibody fragments, to the development of similar cleaving catalysts against the TTR amyloids underlying SSA.
As reviewed in that earlier posting, this approach in principle offers multiple advantages in Alzheimer's disease and brain aging relative to other Aβ immunotherapies, in terms of both efficacy and safety. What Paul now came to realize was that these same features, translated into a catalytic TTR-targeting immunotherapy, would in principle overcome all the obvious limits to those currently in development. They would remove existing aggregates, rather than retarding their further accumulation; exploitation of Paul's protocols would allow them to enrich for highly specific antibody fragments with high catalytic activity; they are catalytic in their activity, reducing the quantity and frequency of injected antibody required to achieve aggregate clearance; they hydrolyze the aggregates, rather than simply sequestering them for later passive degradation or excretion, and do not form stable antibody-Aβ intermediates, avoiding potential side-effects of monomer mobilization and interaction of complexes with surrounding tissues; and they do not rely on patients' immune response for efficacy, eliminating the major possible cause of reduced efficacy in the biologically aged patient and the risk-fraught necessity to induce a strong inflammatory response for their effectiveness.(9)
Listening to Paul, Primmer quickly recognized the high potential of this approach. But it would be difficult to identify suitable patients for clinical trials, or to ensure that even an approved therapy would reach the patients who need it most, if there remained no effective, noninvasive, in vivo diagnostic method for SSA, especially at relatively early ages. By happy coincidence, Paul was already scheduled to speak at the first International Forum on Immunoglobulin Research at Fort Lauderdale, FL that November, as was Dr. Brian O’Nuallain. Primmer was already familiar with the work of Drs. Alan Solomon and O’Nuallain on TTR-targeting antibodies, and the three arranged to meet at the conference, to discuss the possibility of a collaboration. The product of these conversations was a draft proposal for a four-phase research and development project:
Phase 1 will consist of in vitro generation of two separate sets of monoclonal antibodies: (a) antibodies that bind to TTR amyloid for subsequent diagnostic use, and (b) catalytic antibodies that can directly destroy TTR amyloid. ... Phase 2 will consist of studies in an animal model of TTR amyloidosis to determine the safety and efficacy of the potential diagnostic and therapeutic antibodies discovered in Phase 1. Phase 3 will involve clinical trials on the diagnostic and therapeutic potential of the novel antibodies for comparatively young human subjects who develop amyloidosis due to mutation(s) in the TTR molecule. Initial testing of the methodology in the younger cohort is designed to avoid risk to the uniquely fragile group consisting of supercentenarians. Phase 4 will then serve to apply the results of previous research to volunteer supercentenarians in order to improve their health and extend their lives beyond what they would otherwise be expected to live.(4)
The initial phases of the project would be divided according to the expertise of the respective researchers, with O'Nuallain largely responsible for the initial development of a diagnostic binding antibody against wild-type TTR aggregates and later testing in his existing animal models, and Paul for the isolation, purification, and pharmacological optimization of the catalytic cleavage antibody fragment. Along with the proposal came anticipated research budgets for the first two phases, with estimates for the remaining phases to be developed based on their fruits.
The critical issue, of course, was securing funding for such a novel project on an underprioritized age-related disease. Aware of SENS Foundation's recognition of the critical-path priority of such research in the development of comprehensive rejuvenation biotechnology, Primmer submitted the preliminary proposal to CSO de Grey. Following further clarification and negotiation of the draft protocol, and consultation with SENS Foundation's Research Committee, all parties agreed upon a final research plan for Phase I, and the $150 000 research investment was approved by SENS Foundation and disbursed to the researchers.
We are delighted to have such a strong project underway, in the hands of recognized experts in the field, and I am personally grateful for the critical networking done by Primmer and the Supercentenarian Research Foundation to bring the proposal together, and proud of SENS Foundations role as funder of this promising project. TTR-based senile cardiac amyloidosis is an underdiagnosed and heretofore-untreatable disease that is prevalent in the aging population and poised to become a widespread medical problem with the aging of the global population and the improved treatment of more familiar age-related diseases. We will watch the progress of this latest funded research project with keen anticipation, and look cautiously forward to the ultimate culmination of the preclinical groundwork in a therapy for human patients.
References
1. Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
2. Kholová I, Niessen HW. Amyloid in the cardiovascular system: a review. J Clin Pathol. 2005 Feb;58(2):125-33. Review. PubMed PMID: 15677530; PubMed Central PMCID: PMC1770576.
3. Steiner I, Hájková P. Patterns of isolated atrial amyloid: a study of 100 hearts on autopsy. Cardiovasc Pathol. 2006 Sep-Oct;15(5):287-90.
4. Primmer SR, Paul S, O’Nuallain B. Research project to extend lives of supercentenarians by diagnosing and treating transthyretin amyloidosis. Unpublished MS, Supercentenarian Research Foundation. 2010 Mar 25.
5. Hrncic R, Wall J, Wolfenbarger DA, Murphy CL, Schell M, Weiss DT, Solomon A. Antibody-mediated resolution of light chain-associated amyloid deposits. Am J Pathol. 2000 Oct;157(4):1239-46. PubMed PMID: 11021828; PubMed Central PMCID: PMC1850152.
6. Terazaki H, Ando Y, Fernandes R, Yamamura K, Maeda S, Saraiva MJ. Immunization in familial amyloidotic polyneuropathy: counteracting deposition by immunization with a Y78F TTR mutant. Lab Invest. 2006 Jan;86(1):23-31. PubMed PMID: 16357867.
7. Goldsteins G, Persson H, Andersson K, Olofsson A, Dacklin I, Edvinsson A, Saraiva MJ, Lundgren E. Exposure of cryptic epitopes on transthyretin only in amyloid and in amyloidogenic mutants. Proc Natl Acad Sci U S A. 1999 Mar 16;96(6):3108-13. PubMed PMID: 10077645; PubMed Central PMCID: PMC15903.
8. Costa PM, Teixeira A, Saraiva MJ, Costa PP. Immunoassay for transthyretin variants associated with amyloid neuropathy. Scand J Immunol. 1993 Aug;38(2):177-82. PubMed PMID: 8394031.
9. Paul S, Planque S, Nishiyama Y. Beneficial catalytic immunity to Abeta peptide. Rejuvenation Res. 2010 Apr-Jun;13(2-3):179-87. Review. PubMed PMID: 20370602.
10. Lutz W, Sanderson W, Scherbov S. The coming acceleration of global population ageing. Nature. 2008 Feb 7;451(7179):716-9.
Posted 23 September 2010 - 01:50 PM
This October, Hannover and Detroit will host two of the year's most interesting and wide-ranging scientific conferences in the biomedical field. I'll be chairing sessions at both events, focused on the application of regenerative medicine to aging and aging-related disease - a synergy we at SENS Foundation term rejuvenation biotechnology.
<!--break-->
The World Stem Cell Summit - hosted this year in Detroit, Michigan from October 4th to 6th - is a wide-ranging event covering topics from basic research to social policy and ethics, and expected this year to attract more than 1,200 delegates from 30 nations.
I'll be chairing a session at the summit entitled "Regenerative Medicine Against Aging—Technological, Political and Commercial Obstacles and Opportunities". Participants include Dan Perry, of the Alliance for Aging Research; Michael West, acclaimed biotechnology entrepreneur and CEO of Biotime, Inc.; and Huber Warner, former associate director of the National Institute on Aging.

The World Congress on Preventive & Regenerative Medicine, hosted this year in Hannover from October 5th-7th, is the only international event addressing the entire regenerative medicine sector. This broad remit, similar to that of the SENS conference series, gives the meeting outstanding potential to foster interdisciplinary collaborations in research and development.
I am serving as a vice-president of the Congress, and will co-chair a session entitled "Rejuvenation Biotechnologies: Applying Regenerative Medicine to Aging".
I hope to see you at one or both of these exciting events!
Posted 09 October 2010 - 06:25 PM
Neurofibrillary tangles (NFT -- cytoplasmic inclusions composed of phosphorylated and abnormally-cleaved species of tau protein) accumulate in the aging brain, and at higher levels in Alzheimer's disease and in vulnerable regions in a range of other neurodegenerative diseases; they are closely associated with neuronal death and with onset of clinical dementing disease. The clearance of neurofibrillary tangles and other intracellular aggregates from the aging brain is a key rejuvenation biotechnology to restore aging brain function.
The priority of a distinct therapy for the removal of tau pathology has become especially clear in light of followup studies in persons receiving the original, active beta-amyloid vaccine AN1792. On the one hand, vaccine responders' brains exhibited a nearly complete absence of Aß pathology at autopsy, along with reduced neuronal loss, and in long-term (4.6 y) followup, a decline on the Disability Assessment for Dementia and Dependence Scale, stabilized hippocampal volume while adjuvant-only controls suffered ongoing declines, and extensive clearance of tau-containing neurites.(1-3,9) Yet narrowly cognitive benefits were limited, and more mature tau pathology (NFT and neuropil threads) appeared to be unaffected ((1-3), and see previous postings).This last finding, combined with the stronger association of NFT burden with clinical disease, recommend NFT clearance as a high-priority (and, ideally, complementary) immunotherapeutic appro
In previous posts, we highlighted work by Dr. Einar M Sigurdsson and colleagues in targeting tau aggregates in animal models, demonstrating significant reductions in tau pathology and functional deficits. Now, Dr. Hanna Rosenmann's group at Hadassah University Hospital, Israel, has reported even more impressive results, in a rodent model more closely reflecting human age-related neurodegenerative disease.
Rosenmann's group were the first to report the presence of endogenous anti–NFT antibodies in the healthy elderly and AD patients, with the latter group exhibiting a prominent IgM isotype,(4) consistent with the feasibility of NFT-targeting immunotherapy, but also suggesting the possibility of an NFT-targeting immune mechanism in the pathogenesis of AD. In previous work (2) intended primarily to test the latter hypothesis, vaccination of transgenic tau mice with full-length tau potein "induced histopathologic features of Alzheimer disease and tauopathies, indicated by the presence of neurofibrillary tangle-like structures, axonal damage, and gliosis ... [and] mononuclear infiltrates without demyelination in the central nervous system, accompanied by neurologic deficits".(5) Their new work (6) was therefore an effort to successfully and safely clear tau pathology using a vaccination approach.
The investigators used two distinct tau-TG mouse models. The first, a relatively uncomplicated model, involved mice hemizygous for two tau mutant mutations associated with human disease: K257T and P301S (DM-Tau-tg). These mice begin to develop tau pathology at 6 months of age, along with severe deficits in synaptic plasticity and long-term potentiation (LTP). the second, "enhanced" model sought to both accelerate the development of NFT and to increase the likelihood of observing any risk from their new vaccine protocol by better modeling the inflammatory environment of the AD brain, by inducing experimental autoimmune encephalomyelitis (EAE, an protocol to induce brain inflammation, which is generally used as a model of multiple sclerosis) at 6–7 weeks of age.
These aspects of the protocol reflected the design of their original, worrisome study.(5) In that study, however, the animals had been immunized with full-length, wild-type recombinant human tau protein, whereas in this study (6) the immunogen was the pathological, NFT-related phosphorylated tau (phos-tau) species.
Following vaccination, anti-phos-tau antibodies were detected in serum and CNS blood vessels:"(6)
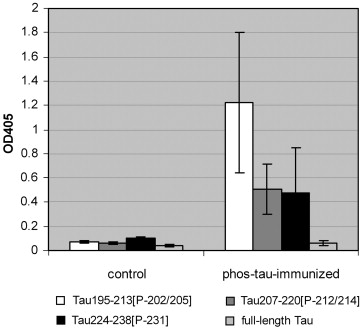
Anti-phos-tau Antibodies in Serum and CNS of Vaccinated Mice.(6)
More impressively, CNS NFT burden was reduced by ~40% following vaccination:
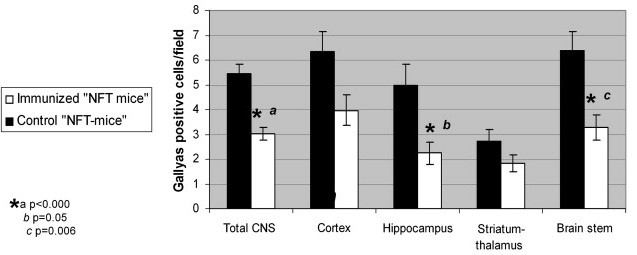
NFT Clearance from CNS of Vaccinated Mice.(6)
Importantly, while these effects were accompanied by an increase in microglial burden (which, in fact, they and others had also found to be correlated with NFT load in human tauopathies and animal models), here reassuringly the activation state of these microglia was no different from that of the pre-existing microglia in NFT-model mice, with or without the inflammatory PT "accelerant". Moreover, "no encephalitogenicity (free of clinical neurological deficits, of adverse effects on brain inflammatory cells and of axonal damage) was recorded." And "unlike the increase in microglial burden the astrocytes were not affected by the phos-tau immunization.
There was also evidence, consistent with results from Sigurdson's group, of both direct antibody interaction with intraneuronal tau aggregates, and of lysosomal involvement in the clearance of the targeted species:
The level of the lysosomal proteases, cathepsins D and L, was affected in the immunized mice ... [But] unexpectedly this was a decrease rather than an increase. The indication for a response of the lysosomal system, together with the presence of Igs in CNS blood vessels ... [suggests that] Anti-phos-tau Abs from the periphery may reach the CNS and enter the neurons either via surface receptors ... or to some extent in association with membrane-bound tau or maybe by direct translocation. The Abs may then bind to intracellular phos-tau aggregates to form anti-phos-tau Abs/phos-tau complexes. Formation of such Ab-Ag complexes can be predicted from our results showing the ability of the serum from immunized mice to recognize cell structures in the CNS of the NFT mice.(6)
While phos-tau-targeting Ab interaction with intraneuronal tau aggregates is initially surprising, it could potentially be enabled by the localization of tau protein to the plasma membrane. Moreover, as recently reviewed by Wisniewski and Siggurdson,
such an outcome is supported by a study of immunization in a Parkinson's disease transgenic mouse model with α-synuclein showing a reduction of intracellular α-synuclein aggregates. Another study has shown that antibodies against Aβ can be internalized in AD neuronal culture models of Aβ accumulation and clear intraneuronal Aβ aggregates via the endosomal–lysosomal pathway [work in Gunnar Gouras' laboratory, reviewed in an earlier post]. Furthermore, recent evidence has shown that extracellular tau aggregates can be internalized and promote the fibrillization of intracellular full-length tau in a tissue culture model. In addition, injection of fibrillar tau brain extract into the brains of transgenic wild-type expressing mice can induce the formation of human tau into filaments, as well as the spread of pathology from the site of injection into neighboring brain regions. This type of “infectivity” of abnormal protein conformation from outside the cell has also been demonstrated for polyglutamine aggregates and is well characterized in prion disease. Aβ has also been shown to have such “infectious” properties in vivo, being able to induce an acceleration of both further Aβ and tau-related pathology. Hence, if the spread of [paired tau helical fragment] pathology in AD can occur via such a prion-like mechanism, antiphosphorylated tau antibodies would not need to enter cells to be effective.(10)
As to the paradoxical reduction in cathepsins D and L,
recent studies did show that inhibition of the lysosomal system can indeed induce degradation of tau ([citing (7) below] mediated via activation of the non-lysosomal protease, calpain ([(8) below]) ... Additional studies are certainly needed to clarify ... this possible “tau degradation induced by lysosomal inhibition” mechanism. ... . The suggested bidirectional role of the lysosomal system (once activated and once inhibited) in the degradation of tau may also be supported by the capability of cathepsin D not only to eliminate tau as part of activated lysosomes but also to proteolyse tau to fragments favouring tangle formation and to enhance its phosphorylation, especially when the cathepsin is released from the lysosomes and reaches the cytoplasm ...
Finally, their responsibly-optimistic conclusion:
[Sigurddson's previous work] used a different peptide immunogen and protocol (more tolerated), demonstrating its efficacy in reducing NFTs. We demonstrate here, in addition to the efficacy of our three phos-tau peptide immunogens against the NFT pathology, that our vaccine, tested under conditions aimed to detect neurotoxicity, is free of adverse effects.
The robust anti-NFT effect and the lack of encephalitogenicity in NFT mice immunized with phos-tau peptides, even though CFA with PT was included in vaccine, point to their anti-NFT therapeutic potential.(6)
These are indeed promising findings, and a clear advance in this key element of a comprehensive panel of rejuvenation biotechnologies, particularly but not exclusively for the preservation and restoration of youthful brain structure function.
References
1. Rodrigue KM, Kennedy KM, Park DC. Beta-amyloid deposition and the aging brain. Neuropsychol Rev. 2009 Dec;19(4):436-50. Epub 2009 Nov 12. Review. PubMed PMID: 19908146; PubMed Central PMCID: PMC2844114.
2. Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
3. Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005 Jan 11;64(1):129-31. PMID: 15642916 [PubMed - indexed for MEDLINE]
4: Rosenmann H, Meiner Z, Geylis V, Abramsky O, Steinitz M. Detection of circulating antibodies against tau protein in its unphosphorylated and in its neurofibrillary tangles-related phosphorylated state in Alzheimer's disease and healthy subjects. Neurosci Lett. 2006 Dec 20;410(2):90-3. PubMed PMID: 17095156.
5: Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006 Oct;63(10):1459-67. PubMed PMID: 17030663.
6: Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010 Aug;224(2):472-85. Epub 2010 May 28. PubMed PMID: 20546729.
7: Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009 Nov 1;18(21):4153-70. Epub 2009 Aug 4. PubMed PMID: 19654187; PubMed Central PMCID: PMC2758146.
8: Zhang JY, Peng C, Shi H, Wang S, Wang Q, Wang JZ. Inhibition of autophagy causes tau proteolysis by activating calpain in rat brain. J Alzheimers Dis. 2009 Jan;16(1):39-47. PubMed PMID: 19158420.
9. Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, Tompkins C, Leibman C, Pomfret M, Grundman M; AN1792 (QS-21)-251 Study Team. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. 2009 Apr;6(2):144-51. PubMed PMID: 19355849; PubMed Central PMCID: PMC2825665.
10: Wisniewski T, Sigurdsson EM. Murine models of Alzheimer's disease and their use in developing immunotherapies. Biochim Biophys Acta. 2010 Oct;1802(10):847-59. Epub 2010 May 13. PubMed PMID: 20471477; PubMed Central PMCID: PMC2930136.
Posted 19 October 2010 - 09:16 AM
In September 2007 SENS Foundation's Chief Science Officer, Dr Aubrey de Grey, together with co-author Michael Rae published Ending Aging - an accessible description of the SENS platform. The book speaks to a broad audience, without "dumbing down" the science in any way, and is thus the ideal resource for both biologists and non-biologists who want to learn more about SENS and its implications.
Science moves quickly, however, especially in a field as new and dynamic as regenerative medicine. A year after its initial publication, Ending Aging was re-released in paperback format with a new afterword, explaining how cutting-edge research since the publication of the hardback had impacted on our plans and expectations - and introducing a detailed model that predicts the tremendous benefits to be expected from rejuvenation biotechnology.
We've been conscious since the re-release that those who had already purchased the hardback would be likely to miss out on this extra information. Consequently, we're delighted to now be able to make the afterword available online for free download.
Posted 20 October 2010 - 03:44 PM
SENS Foundation's CSO, Dr Aubrey de Grey, was first featured in Wired magazine in 2008, shortly before the Methuselah Foundation's initial USA-based conferences. Two years on, he's now returned in a more candid and detailed interview, discussing how the tactics involved in bringing rejuvenation biotechnologies to the attention of mainstream science have evolved and begun to bear fruit.
One of the key arrows in that quiver has been the establishment of SENS Foundation as an independent entity (a decision which Dr de Grey explains at length in the interview), and we're sure that all of our supporters will enjoy reading Aubrey's account of his take on the last couple of years.
Posted 22 October 2010 - 01:28 AM
As noted in a previous post, the clearance of neurofibrillary tangles (NFT) and other intracellular aggregates is a key rejuvenation biotechnology to restore youthful function to aging brains, especially those with Alzheimer's disease (AD) and a range of other age-related neurodegenerative disorders. While Phase I and II clinical trials with the original, active beta-amyloid (Aβ) vaccine AN1792 have demonstrated a wide range of benefits in patients exhibiting a sufficiently robust immunological response, autopsy studies in such patients have reported no evident effect on mature tau neuropathology (NFT and neuropil threads); this seems likely to be a significant barrier to the the potential benefits of Aβ clearance, consistent with the limited cognitive benefits observed in these trials.
However, as we discussed in an earlier post, a recent study(1) did find, amongst other things, a substantial restoration of normal curvature in immunized patients' neuronal processes, accompanied by significantly fewer hippocampal neurons stained with paired helical fragment-1 (PHF1) antibodies, which target a range of hyperphosphorylated tau (phospho-tau) species. These effects were not, however, accompanied by parallel reductions in staining by Alz50 antibodies (recognizing tau phosphorylated at serines 199 and 202 and threonine 205) or Thioflavin-S, suggesting that AN1792 can remove early-stage, but not more mature phospho-tau aggregates.
While suggestive, this study(1) was limited by a small patient base (n = 5) and minimal therapeutic exposure: the Phase II trial in which they had participated was halted due to side-effects (meningoencephalopathy in ~6% of immunized patients) after patients had received only two doses of AN1792. Now, a new study has more strongly reinforced and expanded this finding.
The new report(2) compared neuropathology at autopsy in 10 AD subjects (iAD) immunized with AN1792 in the earlier Phase I trial, to 28 untreated control AD (cAD) subjects. Unlike the subjects in the earlier report (1), these subjects had received 5-7 sessions of vaccine over the course of treatment.
The phospho-tau load was lower in the iAD than the cAD group in the cerebral cortex (cAD 1.08% vs. iAD 0.72%, P = 0.048), CA1 hippocampus (cAD 2.26% vs. iAD 1.05%; P = 0.001), subiculum (cAD 1.60% vs. iAD 0.31%; P = 0.001) and entorhinal cortex (cAD 1.10% vs. iAD 0.18%; P < 0.001). ... Aβ immunotherapy-associated reduction was confined to neuronal processes, i.e. neuropil threads and dystrophic neurites. ... There was a significant correlation between phospho-tau dystrophic neurite cluster counts and Aβ42 load in the cortex (r = 0.879, P = 0.001) but not with phospho-tau load or phospho-tau-positive neurons. This correlation is consistent with the localisation of dystrophic neurite clusters to plaques. ... However, the phospho-tau accumulation in the neuronal cell bodies, contributing to neurofibrillary tangles, appeared not to be affected. ...
Figure 1. Examples of "the cerebral neocortex (a) and CA1 hippocampus (b) of Aβ42 (21F12 antibody) versus phospho-tau (AT8 antibody) staining of an unimmunised AD control (cAD) and an immunised AD case (iAD).From (2).
In probing the mechanism of clearance of early tau pathology in dystrophic neurites, the authors note the possibility that this could be due to the actual phagocytic removal of the phospho-tau-laden dystrophic neurites themselves. Their findings suggest otherwise, however, as
we have not observed any evidence of phospho-tau within microglia, whereas Aβ is readily detectable within microglia in many of these immunised AD cases. This observation may be interpreted as suggesting that the phospho-tau is not cleared by phagocytosis. ...
An alternative explanation is that the neurons are able to mobilise the aggregated tau in the dystrophic neurites and remodel the neurites. The mechanism of clearance of phospho-tau is difficult to address directly in a study of this type. ... However, experimental models with aggregation of both Aβ and tau do exist. In triple transgenic (3 × Tg-AD) mice early, but not the late, forms of phosphorylated human tau were removed by a single injection of anti-Aβ antibody into the brain [3]; reaccumulation of Aβ preceded reappearance of neuronal tau. In another model, active Aβ immunisation of APPSw/NOS2−/− mice, which develop Aβ deposits as well as hyperphosphorylated mouse tau, produced a significant reduction of deposits of both Aβ and hyperphosphorylated tau[4]. Our findings in human AD following Aβ immunisation are consistent with these experimental observations linking Aβ and tau pathobiology. ... This is consistent with the amyloid cascade hypothesis which states that tau pathology is linked to and downstream from Aβ accumulation in AD and confirms the position of Aβ as a target for modifying both Aβ and tau aggregation in AD. (2)
Indeed, recent studies(notably (5-7)) would seem to highlight an even more optimistic reading of the specific clearance of phospho-tau from neuronal processes. In a variety of systems, these reports suggest that Aβ-associated neurotoxicity and synaptic dysfunction may in large part be mediated by aberrant redirection of tau from its normal axonal localization to the somatodendritic compartment, even in the absence of any change in the total levels of tau or of axonal transport proteins. Importantly, lowering tau protein levels affords significant protection against such toxic effects, despite a lack of effect on baseline axonal transport. This is consistent with many revious studies (eg, (8-10)) showing that depletion of native or transgenic tau protects rodent models of AD against Aβ neurotoxicity, even absent an effect on NFT These findings therefore suggest that the ability of Aβ immunotherapy to clear early tau pathology from the dentrites, as found in the current report,(2) may have an unexpectedly powerful neuroprotective effect, if initiated at a relatively early stage in the "amyloid cascade," even if not accompanied by concomitant NFT clearance. Additionally, it seems reasonable to expect that the accumulation of NFT may be substantially prevented by such early immunotherapeutic clearance of Aβ, accompanied by clearance of its precursors in the dendrites.
The new report does however seem consistent with a much more limited benefit of Aβ immunotherapy alone once the disease is well-esatablished, as in these patients and those in earlier autopsy reports:
One possible explanation for the continued decline in function [in AD patients after Aβ immunisation] may be the lack of protective effect of Aβ immunisation on accumulation of phospho-tau in the neuronal cell bodies, implying that additional therapy may need to be directed at neurofibrillary tangles. ... Each of the iAD patients had cognitive function scores within the range MMSE score 15–25/30 at the time of their immunisation. This MMSE range is associated with Braak stages III–V whereas Braak stage VI is associated with MMSE scores in the range 0–10. The decline in MMSE score to 0 in all but one of the iAD cases prior to their death is therefore in keeping with progression in their Braak stage following Aβ immunisation. Correspondingly, the lack of difference in phospho-tau-positive neuron counts between the iAD and cAD cases suggests that phospho-tau continued to accumulate in neuronal cell bodies to reach, by the time they died, a level indistinguishable from that in unimmunised terminal AD. We cannot exclude an upstream role of smaller tau aggregates (not detected by AT8 staining) in the cognitive deterioration as observed in animal studies
In showing that Aβ immunisation can influence phospho-tau pathology, we confirm the position of Aβ as a target for modifying tau accumulation in AD and demonstrate a link between these proteins. However, the continuing progression of cognitive decline in AD patients after Aβ immunisation may be explained by its lack of apparent effect on tangles.(2)
As the authors note, and as animal studies reviewed in an earlier post suggest, immunotherapy directed at clearance of tau pathology would seem to offer a way to overcome this limitation.
References
1. Serrano-Pozo A, William CM, Ferrer I, Uro-Coste E, Delisle MB, Maurage CA, Hock C, Nitsch RM, Masliah E, Growdon JH, Frosch MP, Hyman BT. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010 May;133(Pt 5):1312-27. Epub 2010 Mar 31. PubMed PMID: 20360050; PubMed Central PMCID: PMC2859150.
2. Boche D, Donald J, Love S, Harris S, Neal JW, Holmes C, Nicoll JA. Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer's disease. Acta Neuropathol. 2010 Jul;120(1):13-20. Epub 2010 Jun 9. PubMed PMID: 20532897.
3. Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004 Aug 5;43(3):321-32. PubMed PMID: 15294141.
4. Wilcock DM, Gharkholonarehe N, Van Nostrand WE, Davis J, Vitek MP, Colton CA. Amyloid reduction by amyloid-beta vaccination also reduces mouse tau pathology and protects from neuron loss in two mouse models of Alzheimer's disease. J Neurosci. 2009 Jun 24;29(25):7957-65. Erratum in: J Neurosci. 2010 Jan 20;30(3):1197-8. PubMed PMID: 19553436; PubMed Central PMCID: PMC2871319.
5. Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010 Oct 8;330(6001):198. Epub 2010 Sep 9. PubMed PMID: 20829454.
6. Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010 Sep 8;30(36):11938-50. PubMed PMID: 20826658.
7. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010 Aug 6;142(3):387-97. Epub 2010 Jul 22. PubMed PMID: 20655099.
8. Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007 May 4;316(5825):750-4. PubMed PMID: 17478722.
9. Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006 Dec 22;281(51):39413-23. Epub 2006 Oct 20. PubMed PMID: 17056594.
10. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005 Jul 15;309(5733):476-81. PubMed PMID: 16020737; PubMed Central PMCID: PMC1574647.
Posted 28 October 2010 - 12:50 AM
The intrinsic biological aging process is driven by the accumulation of damage to the cellular and molecular structures in tissues and organs, resulting from the biochemical side-effects of essential metabolic processes. In turn, this rising decay of cellular and molecular structures drives the age-related rise in disability, disease, dependence, dementia, and ultimate risk of death. Because of this well-established connection, and because of the methodological difficulties in evaluating the effects of interventions on the extension of the youthful health and functionality of model organisms, the ability of an intervention to extend life in well-husbanded, nonobese, longevous strains of laboratory animals remains a necessary if surrogate metric for evaluating the potential of interventions to maintain or restore the health of aging organisms, including humans.
To date, very few interventions have been found to meet this rigorous standard -- and most of them involve germline mutations, of no likely human translatability. The most robustly-documented environmental manipulation that extends life and health in mammals remains Calorie restriction (CR), and this has led to a strong interest in the biogerontology community in evaluating pharmacological agents that might might provide its benefits without requiring the arduous adoption of a CR diet in humans -- so-called "CR mimetics."(1) In an earlier post, we reviewed a long list of putative CR mimetics that have failed in lifespan studies. Amongst these was the phytoalexin polyphenol resveratrol, famously found in trace amounts in wine and widely anticipated to be one of the first effective life-extending, youth-preserving compound, but found ineffective in testing in nonobese, longevous mice.(2) We also reviewed the results of the serendipitous late-life lifespan study of rapamycin (sirolimus/Rapamune®), an inhibitor of the mammalian Target of Rapamycin (mTOR) pathway, through the NIA's Interventions Testing Program (ITP), "a multi-institutional study investigating treatments with the potential to extend lifespan and delay disease and dysfunction in mice." This study was hailed as a breakthrough, being the first robust demonstration of lifespan extension in mammals by a pharmacological agent, although as we reviewed, the absolute effect of rapamycin was limited, and in fact not entirely clear in males.(3)
The ITP has now provided important confirmation of these findings, by testing resveratrol at higher doses using more robust animal models, and rapamycin beginning in much younger adult mice.
Rapamycin was administered in food to genetically heterogeneous mice from the age of 9 months and produced significant increases in life span, including maximum life span, at each of three test sites. ... Rapamycin was found to lead to improved survival in both males and females when pooling across test sites, and to significant effects at each test site considered separately. ... For males, rapamycin led to an increase of 10% in median age, averaged across the three sites, and an increase of 16% in the 90th percentile age [ie, by operational definition, maximum lifespan -MR]. For females, the corresponding values were 18% for median, and 13% for 90th percentile ages. ... Rapamycin attenuated age-associated decline in spontaneous activity in males but not in females. Causes of death were similar in control and rapamycin-treated mice.(4)
The results are important on several fronts. The activity test gives some preliminary additional evidence of extended "healthspan" in rapamycin-administered mice. Similarly, the lack of a differential effect on specific causes of death suggests again that the life extension effects of rapamycin are the result of a far-reaching improvement in all the animals' systems, and not due to inhibiting some single, and perhaps idiosyncratic, overriding cause of death. The confirmation of the effect in males was important, as it had been ambiguous in the earlier study, thanks to a surprising trend toward an early survival advantage in treated males prior to the onset of intervention, and the lack of any clear effect at one of the 3 testing centers (The Jackson Laboratories).
The actual lifespan results, however, were also revealing, and in a surprising way:
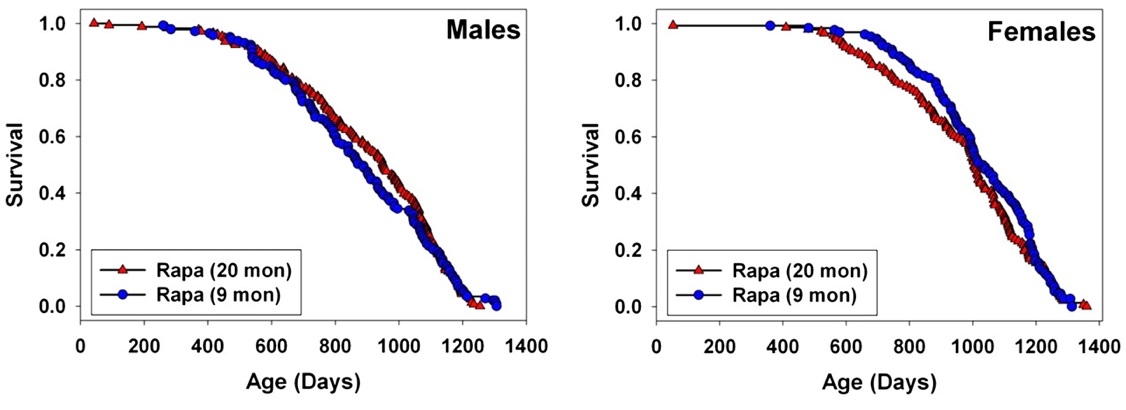
Overlapping survival curves from rapamycin administered in early or late adulthood in mice. From (4)
[This figure] a graphical comparison between the results of the current study [with rapamycin administered from age 9 mo onward (4)], and the results of the study comparing mice exposed to rapamycin from 20 months of age [(3)]. The two data sets were not produced simultaneously, but they do represent work done using the same conditions of drug preparation, diet, water source, housing, and genetic stocks at the same three sites, with only a 1 year lag between start dates. For male mice, starting rapamycin at 9 months rather than at 20 months did not lead to any improvement in survival [relative to the later age of onset]; the two cohorts were not significantly different by log-rank test, at p = .74. For female mice, there is a suggestion that earlier exposure to rapamycin may have led to some slight decline in mortality risk before ∼1000 days of age, but the log-rank test is not significant (p = 0.31). The Wilcoxon–Breslow test, which does not assume that the difference in risk between the test groups is constant at all ages and which gives more weight to earlier deaths, was also nonsignificant for females, at p = 0.09 (two tailed). ... In principle, however, an agent that slowed the overall process of aging would be expected to have a stronger effect if started at a relatively early age. [This is because it would be expected that the greater duration of treatment would prevent a larger net burden of aging damage to accrue, leading to greater lifespan benefit, as is observed eg. in CR administered at weaning, in middle age, and toward the end of the "natural" lifespan (5) -MR] Thus, if later work confirms that rapamycin treatment starting in later life is fully as effective as treatment started early in adult life, this could be taken as evidence that the agent is not modulating aging so much as limiting the pace of, or vulnerability to, late life illnesses, such as the neoplastic diseases that lead to most of the deaths in UM-HET3 mice.(4)
Alternatively, it might suggest that the intervention is not interfering with the underlying structural decay of aging, but in some of its downstream metabolic sequelae, affording treated animals a greater ability to withstand the pathological systemic effects of aging damage once the burden is sufficient to exert a systemic, mortality-accelerating influence. Such an effect was suggested in an earlier, not fully convincing report of similar life extension in mice were administered the spin trap antioxidant, N-tert-butyl-alpha-phenylnitrone (PBN), at ages 18.5, 21.5, or 24.5 mo.(6) This is also consistent with the implications of observations of systemic effects of aging on stem cells(7,8) and of the antioxidant effects of R-lipoic acid when administered to young, but not to old, rodents,(9) and possibly with the effects of cognitive engagement in delaying the onset of clinical dementia but accelerating its course.(10-12)
The confirmation of a lack of effect on lifespan at higher doses of resveratrol was equally important in its own way, granted its high prominence and its ongoing fueling of sales and unscrupulous promotional tactics in the weakly-regulated dietary supplement market:
Resveratrol (at 300 and 1200 ppm food) and simvastatin (12 and 120 ppm) did not have significant effects on survival in male or female mice. ... Our resveratrol data thus serve to confirm the absence of any effect of this agent on mouse life span, using doses two- to eightfold higher than the dose studied by Pearson and colleagues [(2)] and using genetically heterogeneous mice of both sexes rather than male C57BL/6Nia mice alone. (4)
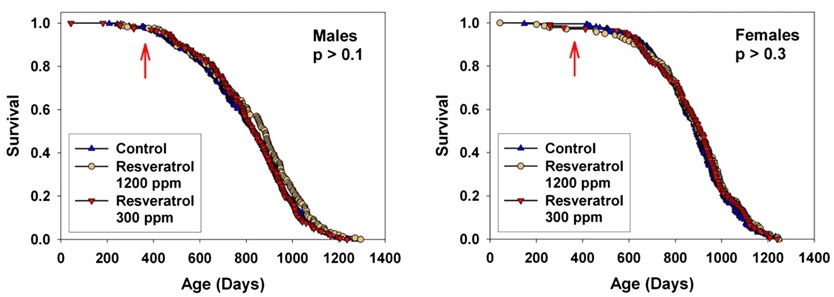
Resveratrol Fails to Extend Life in Normal, Genetically-Heterogeneous Mice. From (4)
This latter point is noteworthy, not only because of the a priori reasoning that a study population with a wider genetic diversity is less likely to lead to artifacts due to a strong effect on some strain-specific, life-limiting disease, but because of the specific concern that the authors of the earlier report had raised:
resveratrol treatment did not significantly alter the distribution of pathologies in [standard diet] groups. This included neoplasias, despite the potency of resveratrol against implanted or chemically induced tumors ... This may be related to the fact that the vast majority of these cases were lymphomas, a tumor type for which the efficacy of resveratrol has not been thoroughly assessed, and that is thought to be triggered mainly by endogenous retroviruses in mice (2)
Combined with their positive results with rapamycin, the failure of resveratrol to extend life using resveratrol in normal mice over a very wide range of doses should reasonably be taken to put the resveratrol "story" to test.
On the other hand, the ability of rapamycin to extend life in these mice has been confirmed, and expanded to a preliminary extent. Naturally, further studies are underway or proposed to elucidate the full nature of these effects:
One such study will evaluate doses of rapamycin both higher and lower than the dose used for the initial longevity studies ... Cross-sectional histopathology will help to identify earlier stages of common age-dependent illnesses, including those that do not typically lead to death ...Studies of multiple age-dependent physiological outcomes will also be included. These ... will help to show whether the life span improvement represents merely a global inhibition of neoplastic disease, or rather an authentic antiaging effect that includes anticancer protection as just one among many consequences. The follow-up studies will also evaluate biochemical changes in multiple tissues, to see which tissues show long-term and short-term inhibition of mTOR and modulation of mTOR-dependent cellular feedback circuitry. ... It is equally plausible that the health benefits of rapamycin principally reflect a change in one or a few cell types, perhaps located in hypothalamus, vascular endothelium, a hormone-producing cell type, or a stem cell compartment, whose beneficial effects on other cell types are independent of the level of mTOR function in the downstream target tissues. Evaluation of rapamycin effects on rodent models of specific diseases is likely to be very informative in this regard. For example, two recent studies have evaluated rapamycin effects on different mouse models of Alzheimer’s disease ... Each study found that rapamycin rescued memory deficits and slowed pathological manifestations in these neurodegenerative models. Analysis of effects on other rodent (and perhaps canine) models of late-life diseases should prove equally informative. It will also be helpful to learn if rapamycin can further improve the longevity benefits produced by specific mutations or diets or other drugs. Researchers have learned much about the biology of aging by studies of caloric restriction and of antiaging mutations, and the advent of effective antiaging drugs should provide additional leverage for new work on the factors that time the coordinated appearance of age-related decline. In addition, work on antiaging interventions holds more promise for the eventual development of protective medicines than approaches that entail modification of germline genes or lifelong compliance with extreme dietary restrictions.(4)
At the same time, whatever these studies may reveal, even the most optimistic reading of these results and an assumption of perfect human translatability is still overshadowed by how limited the results are. Interventions such as rapamycin, which only retard the rate at which aging damage accumulates (or, perhaps, allows the organism to carry on functioning for a longer period of time under its accumulating burden), can only temporarily delay the onset of age-related ill-health, not arrest or reverse it -- and in the case of rapamycin, the first pharmacological agent to extend the lives of otherwise-healthy mammals, its ability to do even this has been found to be limited. Rejuvenation biotechnology offers an alternative approach, and the promise of a life of greatly-extended youthful functionality. References 1: Minor RK, Allard JS, Younts CM, Ward TM, de Cabo R. Dietary interventions to extend life span and health span based on calorie restriction. J Gerontol A Biol Sci Med Sci. 2010 Jul;65(7):695-703. Epub 2010 Apr 6. Review. PubMed PMID: 20371545; PubMed Central PMCID: PMC2884086. 2: Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, Jamieson HA, Zhang Y, Dunn SR, Sharma K, Pleshko N, Woollett LA, Csiszar A, Ikeno Y, Le Couteur D, Elliott PJ, Becker KG, Navas P, Ingram DK, Wolf NS, Ungvari Z, Sinclair DA, de Cabo R. Resveratrol Delays Age-Related Deterioration and Mimics Transcriptional Aspects of Dietary Restriction without Extending Life Span. Cell Metab. 2008 Aug;8(2):157-68. PMID: 18599363 [PubMed - as supplied by publisher] 3: Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009 Jul 16;460(7253):392-5. Epub 2009 Jul 8. PubMed PMID: 19587680; PubMed Central PMCID: PMC2786175. 4: Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, Nadon NL, Strong R. Rapamycin, But Not Resveratrol or Simvastatin, Extends Life Span of Genetically Heterogeneous Mice. J Gerontol A Biol Sci Med Sci. 2010 Oct 25. [Epub ahead of print] PubMed PMID: 20974732. 5: Rae M. It's never too late: calorie restriction is effective in older mammals. Rejuvenation Res. 2004 Spring;7(1):3-8. Review. PubMed PMID: 15256039. 6: Saito K, Yoshioka H, Cutler RG. A spin trap, N-tert-butyl-alpha-phenylnitrone extends the life span of mice. Biosci Biotechnol Biochem. 1998 Apr;62(4):792-4. PubMed PMID: 9614711. 7: Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005 Feb 17;433(7027):760-4. PMID: 15716955 8: Mayack SR, Shadrach JL, Kim FS, Wagers AJ. Systemic signals regulate ageing and rejuvenation of blood stem cell niche. Nature. 2010 Jan 28;463(7280):495-500. PubMed PMID: 20110993. 9: Hagen TM, Ingersoll RT, Lykkesfeldt J, Liu J, Wehr CM, Vinarsky V, Bartholomew JC, Ames AB. ®-alpha-lipoic acid-supplemented old rats have improved mitochondrial function, decreased oxidative damage, and increased metabolic rate. FASEB J. 1999 Feb;13(2):411-8. PubMed PMID: 9973329. 10: Wilson RS, Barnes LL, Aggarwal NT, Boyle PA, Hebert LE, Mendes de Leon CF, Evans DA. Cognitive activity and the cognitive morbidity of Alzheimer disease. Neurology. 2010 Sep 14;75(11):990-6. Epub 2010 Sep 1. PubMed PMID: 20811001; PubMed Central PMCID: PMC2942032. 11: Chaves ML, Camozzato AL, Köhler C, Kaye J. Predictors of the progression of dementia severity in brazilian patients with Alzheimer's disease and vascular dementia. Int J Alzheimers Dis. 2010 Mar 14;2010. pii: 673581. PubMed PMID: 20798750; PubMed Central PMCID: PMC2925083. 12: Scarmeas N, Albert SM, Manly JJ, Stern Y. Education and rates of cognitive decline in incident Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2006 Mar;77(3):308-16. PubMed PMID: 16484637; PubMed Central PMCID: PMC2077720.
Posted 09 December 2010 - 06:09 AM
Last night, Peter Thiel hosted "Breakthrough Philanthropy", a dinner and presentation event showcasing eight non-profit organizations focused upon game-changing approaches to their fields. It was a wonderful opportunity for us to convey our message to a large group of entrepreneurs and philanthropists interested in the genuinely transformative. Our thanks to the Thiel Foundation, and the Seasteading Institute, for their efforts in creating that event. Here are my remarks from the presentation.
Good evening. A man by the name of Frank Fenner died two weeks ago. He was 95 years old. Professor Fenner was at one time the Chairman of a W.H.O. Global Commission, and thirty years ago this Spring he was the man who had the honor of declaring that smallpox had been eradicated from the planet.
It was 25 years before that when Dr. Thomas Francis stood up at the University of Michigan to announce that the Salk polio vaccine was “safe, effective and potent.” Just to remind you of the impact of that – Voice of America reported that day that church bells were ringing across the country before he left the podium.
We’ve gotten very good at conquering infectious diseases. None of you will suffer from polio, or smallpox, or, likely, measles or diptheria. But the truth is, we haven’t extended that kind of success to the problems of aging. You will know someone suffering from cancer, from Alzheimer’s, from Parkinson’s disease. We haven’t yet eradicated a single, major age-related disease. This is despite great advances in therapeutics, despite trillions in research underway around the globe, and despite the brightest minds working in an entire, thirty year long biotech revolution.
Ten years ago, our CSO Aubrey de Grey first suggested … a slightly different revolution. His core idea was a damage repair model called SENS, and a recognition of a simple problem: when medical science focuses upon disease – when we wait for disease to develop – then what we learn is how to chase the pathology – and we haven’t gotten very good at that. But if we look instead at the damage building up as a result of the normal metabolism of being alive, then we can learn to repair that damage before those deadly pathologies develop.
That’s it; that’s all that SENS means: it’s a model that steps away from the expensive and tangled ‘pathology chase’ and focuses instead upon identifying, addressing and removing the damage that is building up inside you; the damage that will eventually cause disease.
It’s a simple statement, but it suggests a complex task. For this revolution to succeed we need to move this way of thinking into the mainstream of medicine. We need to create a new biotech industry. That’s why we created SENS Foundation: to be a credible catalyst for change; to be a public research and outreach organization devoted to the creation of a new field, rejuvenation biotechnology.
The groundwork has been laid. Peter Thiel and other early supporters allowed a handful of visionary researchers to take the first steps in this field a few short years ago. They are now being joined by an increasing number of individuals who believe in the Foundation and its mission.
And, because of that, 2010 has been a big year for us. We’ve expanded our research center in Mountain View. We’ve added new research programs, and we’ve added several new collaborating institutions. Our lysosomal research program, especially, has made great progress in addressing macular degeneration and heart disease. Our Academic Initiative is growing the next generation of researchers. All of these activities are generating quality, peer-reviewed publications.
2011 promises to be bigger still. We have everything in place for ten further important projects, and, if we are successful in fundraising, by mid-2011 we’ll be pursuing at least one research program in every currently recognized category of metabolic damage. What we’re most proud of, though, is that our projects are capturing the imaginations of top tier collaborators in biotech and regenerative medicine. Discussions are underway for a center of excellence at Cambridge University, and at last month’s TEDMED conference, in a joint statement with Wake Forest University, we announced a collaboration with their world-renowned Institute for Regenerative Medicine. We are attracting serious attention from the mainstream medical science community – and that’s exactly what we’re trying to achieve.
A man by the name of Frank Fenner died two weeks ago. He was only 95 years old. Medicine hasn’t eradicated a single, major age-related disease … yet … . You can help SENS Foundation change that. Thank you.
Posted 08 January 2011 - 07:42 PM
As we reviewed in a previous posting on a recent advance in genetic engineering with zinc finger nucleases (ZFNs),
In addition to its widely-anticipated potential to provide highly-effective therapies for genetic disorders, somatic gene therapy is an essential enabling technology for the repair or obviation of several of the cellular and molecular lesions driving age-related disease and dysfunction (notably the accumulations of mutations in mitochondrial and nuclear DNA [including the medium-term obviation of the latter through WILT]). One of the most promising routes to somatic gene therapy is zinc finger nucleases (ZFNs), engineered DNA-binding proteins consisting of a FokI restriction enzyme catalytic core bookmarked into a dimer of zinc finger array DNA binding domains. The choice of zinc finger domains allows the engineer to target twinned 9-18 base-pair sequences in the recipient genome, separated from each other by a (typically) 5-7 base pair spacer. Upon binding, the restriction enzyme dimerizes, creating a double-strand break at the spacer locus; the engineer then takes advantage of the native DNA repair machinery to insert an user-supplied DNA repair template through Non-Homologous End Joining (NHEJ).
Image © Sigma-Aldrich Zinc Finger Nuclease Learning Center
Despite the many advantages of ZFNs, and the landmark 2009 report of their use to generate precision knockout mice,(1) relatively little use has been made of AFNs for either basic science or translational work toward gene therapies. This is principally because of the difficulty and expense of generating novel ZFN binding arrays, whose creation using the standard technique of modular assembly is often unsuccessful due to combinatorial incompatibilities amongst individual "finger" peptides which, in sequence, would otherwise be expected to bind to the targeted DNA sequence: when placed in sequence, such peptides are often found to interfere with one another's DNA-binding capacity.
To overcome these difficulties without recourse to expensive and (some argue) overly-restrictive purchase from patent-holder Sangamo BioSciences (via its licensee, Sigma-Aldrich), a group of researchers centered at Massachusetts General Hospital and the University of Minnesota united to form the Zinc Finger Consortium, to create a new platform for ZFN creation on "open source" principles, "committed to developing resources, software, and other tools for engineering zinc fingers and for performing genome engineering that are robust, user-friendly, and publicly available to the academic scientific community." The first fruits of this collaboration, reported in 2008,(2) was OPEN (Oligomerized Pool ENgineering), "a rapid, publicly available strategy for constructing multifinger arrays." Despite its claimed efficiencies, however, it remained labor-intensive and expensive to establish OPEN methods in a given lab and to generate individual ZFNs, requiring as it did that each site establish its own library of peptide variants and then screen the collection for suitable 'fingers' to reach into each desired target site.
Now, the Massachusetts-Minnesota collaboration has reported that they have developed "context-dependent assembly (CoDA), a platform for engineering ZFNs using only standard cloning techniques or custom DNA synthesis":
With the CoDA approach, three-finger arrays are assembled using N- and C-terminal fingers that have been previously identified in other arrays containing a common middle finger (F2 units) (Fig. 1). CoDA can be implemented by using a large archive consisting of 319 N-terminal-end fingers (F1 units) and 344 C-terminal-end fingers (F3 units) engineered to function well when positioned adjacent to one of 18 fixed F2 units. Thus, in contrast to modular assembly, CoDA does not treat fingers as independent modules but instead explicitly accounts for context-dependent effects between adjacent fingers, thereby increasing the probability that a multifinger array will function well. CoDA is rapid and requires neither specialized expertise nor labor-intensive selections; dozens of multifinger arrays can be constructed in 1–2 weeks or less using standard cloning techniques or commercial DNA synthesis.(3)

Figure 1. Schematic Overview of CoDA Method for Engineering Zinc Finger Nucleases. From (3).
If this is indeed the experience at independent labs -- that they can use CoDA in an essentially off-the-shelf way, to rapidly identify and generate novel ZFNs in-house -- then CoDA promises to greatly increase the power of genetic engineering, putting the unprecedented precision of ZFNs in genetic engineering of mammalian systems into the hands of basic researchers and opening up the potential of new gene therapies for recognized congenital diseases, while establishing an enabling technology that will be essential to future rejuvenation biotechnologies. It will also be a powerful validation of the optimism that many academic scientists and DIYbiohackers have poured into open-source biological and technological research and development. We may all some day owe these scientists a great debt, even if -- especially if -- we ultimately rarely recall their names or their report because the technology is so well-established as to be taken for granted, a part of the biomedical background as invisible to future generations that reap its benefits in health and longevity as sanitation systems and the absence of endemic polio are to the developed world today.
References
1. Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, Vincent A, Lam S, Michalkiewicz M, Schilling R, Foeckler J, Kalloway S, Weiler H, Ménoret S, Anegon I, Davis GD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jacob HJ, Buelow R. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009 Jul 24;325(5939):433. PubMed PMID: 19628861; PubMed Central PMCID: PMC2831805.
2. Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, Unger-Wallace E, Sander JD, Müller-Lerch F, Fu F, Pearlberg J, Göbel C, Dassie JP, Pruett-Miller SM, Porteus MH, Sgroi DC, Iafrate AJ, Dobbs D, McCray PB Jr, Cathomen T, Voytas DF, Joung JK. Rapid "open-source" engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008 Jul 25;31(2):294-301. PubMed PMID: 18657511; PubMed Central PMCID: PMC2535758.
3. Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, Curtin SJ, Blackburn JS, Thibodeau-Beganny S, Qi Y, Pierick CJ, Hoffman E, Maeder ML, Khayter C, Reyon D, Dobbs D, Langenau DM, Stupar RM, Giraldez AJ, Voytas DF, Peterson RT, Yeh JR, Joung JK. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods. 2011 Jan;8(1):67-9. Epub 2010 Dec 12. PubMed PMID: 21151135.
Posted 24 January 2011 - 02:05 PM
We've now reintroduced an FAQ page to the SENS Research area of our site, addressing several common queries and concerns about SENS Foundation and our work. If you'd like to suggest a question to be added to the FAQ, please email the webteam.
Posted 01 February 2011 - 10:34 PM
The degenerative aging of the immune system is responsible for an enormous burden of disease and disability, from the pain of recurrent Herpes zoster and postherpetic neuralgia, to elevated rates of chronic urinary tract infections, to complications in wounds, pressure sores, ulcers, and surgical incisions. Most prominently, it underlies the meteoric rise in mortality from respiratory infections with age: influenza, pneumonia, and septicemia rise from being negligible causes of death in healthy middle-aged adults in the USA, to emerge amongst the top 10 causes of death in adults over the age of 55, with mortality rates climbing with each successive year of aging. And in addition to increasing the morbidity and mortality specifically attributable to particular infections, the dysregulation of immune function by immunosenescence is widely acknowledged to exacerbate multiple chronic age-related illnesses, and to contribute to functional decline and frailty in aging people.(1)
While vaccine manufacturers and public health officials have rightly advocated for expansion of population vaccine coverage as a measure to blunt the burden of infectious disease in the elderly, the effectiveness of this strategy is itself limited by immunosenescence, which progressively diminishes the adaptive immune system's response to vaccination with age:
[Preventive] elimination of clinical disease is often unrealistic for older adult populations with diminished immunity and impaired vaccine responses. The goals of immunization in older adults are to prevent serious illness, hospitalization, and death, but benefits relating to exacerbation of underlying chronic illness, functional decline, and frailty are other worthy endpoints ... These endpoints are more difficult to measure and harder to specifically attribute to the organism(s) targeted by vaccines, often leading to conflicting evidence of vaccine efficacy in older adults. For example, although some investigators have found that administering the influenza vaccine to community-dwelling individuals aged 65 and older leads to significant reductions in risk of hospitalization for pneumonia or influenza and death, others have documented little or no effect, particularly in those aged 70 and older with significant comorbidities.(1)
The solution to age-related suffering and death from specific infections, autoimmunity, and inflammation is the application of rejuvenation biotechnology to the aging immune system itself.
The clearest and longest-established contributor to immune senescence is the decline in adaptive immunity mediated by T lymphocytes,(2) the biomedical remediation of which has therefore been the focus of SENS Foundation's investments in immunological rejuvenation research.(9,10) The existence, nature, and causes of age-related deficits in B-cell structure and function have long been less clear, but emerging evidence has recently led to a consensus of their reality, although the mechanisms have not yet been definitively established.(3) Among the key questions are whether and to what degree this decline in humoral immunity attributable to the degenerative aging process is mediated through the introduction of intrinsic defects to the B-cells themselves, vs. alterations in the systemic environment of the aging body. The strategy for the development of rejuvenation biotechnology for this arm of the aging immune system will be determined by the answers to these questions.
Now, a conceptually simple single experiment performed by Doron Melamed and colleagues at the Technion-Israel Institute of Technology(4) has simultaneously provided powerful evidence for the existence of intrinsic defects in an accumulating population of long-resident B-cells in biologically aged hosts, and for a relatively straightforward intervention to substantially restore youthful humoral immunity in such organisms.
Melamed's group studied the effects of the degenerative aging process on B-cell function using young adult (4 mo) and early-old (20 mo)
C57JBl/6 mice. Several models were used to evaluate the possible role of accumulations of long-extant peripheral B cells with age in suppressing B-cell lymphopoiesis and contributing the the overall levels of cell-intrinsic defects in the aging organism's B-lymphocyte population. One such model was transgenic (TG) mice with an inducible Cre/lox system allowing for conditional knockout of the gene encoding the receptor for B-Cell Activation Factor Receptor (Baff-r). Baff-r is a B-cell activation factor in the tumor necrosis factor (TNF) family whose signaling is essential for the survival of mature B lymphocytes, but is conveniently dispensable for generation of new ones. Activation of the recombinase system allowed the investigators to rapidly deplete animals of B-cell populations that had developed, matured, and aged normally. In additional experiments, the Israeli group further confirmed and expanded their findings in Baff-r-TG mice with similar B-cell depletion studies in aging wiltd-type (WT) mice subjected to depleting antibody mixture, and to the targeting of transgenic human CD20 in hCD20-TG mice.(4)
Restoration of Repressed B-Cell Production
Old and young animals' bone marrows contained similar numbers of B lymphocytes, but the proportions of mature versus precursor and and early B-cells were greatly skewed. In young animals animals, 75% of B-lymphocytes in the bone marrow were newborn and precursor cells (proB (B220+/CD43+/IgM-), preB (B220+/CD43-/IgM-), and immature B (B220lo/IgM+)); on old animals, the proportion of such cells had fallen to just 12%. Induced Baff-r knockout in these animals' B-cells cut the B-cell population in peripheral blood and spleen in half, but following this assault, newly-generated and precursor B-cells began to appear in bone marrow at similar frequencies in old mice as in young ones. These results were reinforce in later studies using another depletion model, which further demonstrated that levels of common lymphoid progenitors and multipotent primitive progenitors were depressed in the bone marrow of old mice, but rapidly recovered to youthful levels following an depletion.(4)
Renewal of Lymphopoeisis
These results suggested that the accumulating population of long-resident B lymphocytes in old animals was playing an important role in the age-related decline in B lymphopoiesis. To test this hypothesis further, the investigators depleted B-lymphocytes directly, using a mixture of antibodies, leading to a >80% depletion of peripheral B-cells without substantially reducing the total numbers of newborn and precursor B-lymphocytes in the periphery or the bone marrow. However, the efficiency of repopulation in old animals remained significantly depressed compared to youthful rates: while young mice largely reconstituted their peripheral blood and spleen B-cells within 5 d of depletion, old animals required >50 d to accomplish the same reconstitution -- a rate similar to that previously reported in old animals subjected to ablation using cyclophosphamide or irradiation. But with successive further rounds of B-cell depletion, the old animals began to reconstitute their B lymphocyte populations at increasingly rapid, more youthful rates. After subjecting aged animals to a first round of B-cell depletion and waiting for peripheral blood B-cell numbers to fall by ≥80%, repopulation time following a second round of depletion was reduced to just 18-30 d, falling further to only 8 d following a third round of depletion -- a rate similar to that required in young animals. Consistent with this, the absolute number of precursor B-cells in old animals progressively increased with each round of depletion, nearly equaling that of young animals by the third round.(4)
Re-Emergence of Naïve B-Lymphocytes
The higher frequencies of early-stage B-cells that appeared in the bone marrow and periphery of old animals after the depletion of long-resident B-cell populations was paralleled by rapid declines in the abnormally high numbers of antigen-experienced PanCD45+/B220lo B-cells that had accumulated in the spleens of aging mice. These cells were rapidly replaced by mature but antigen-naïve PanCD45+/B220+ B-cells.When peripheral B-cells were depleted in later experiments using a transgenic immunoglobulin reporter gene to mark newly-generated B-cells, lymphopoieisis was seen to be restored: the absolute number of B-cells in the bone marrow rapidly increased, and in parallel the frequency of newly-generated B220+ cells in the spleen rose >15-fold, and >90% of the reconstituting B cells were of a newly-generated population. Moreover, the population of mature splenic B-cells had shifted rapidly away from antigen-experienced PanCD45+/B220lo cells to naïve PanCD45+/B220+ cells, generating a repertoire not significantly different from that of young animals bearing the same reporter gene.(4)
Renovated Antibody Production
These studies demonstrated that old mice retain the capacity to perform lymphopoeisis at rates comparable to their youth, and to restore the youthful balance of B-cell subpopulations, and that the removal of accumulations of long-resident peripheral B-cells was sufficient to return these aspects of immunological aging to profiles similar to much younger animals. But the most important question, from the perspective of rejuvenation research, still remained. Would this intervention go beyond phenotypic changes in the lymphopoietic system, to rejuvenate the flagging B-cell function of old, immunosenescent animals?
To answer this key question, the investigators subjected old (22 mo) WT mice to B-cell depletion, and then 70 d later challenged them, along with young controls and untreated old mice, with i.p. NP-CGG (Chicken Gamma Globulin). As shown by ELISA, the NP-targeting IgG1response to antigen exposure was greatly reduced in old as compared with young mice (66.5±19.8 units vs. 347.3±47.7). But prior B-cell depletion substantially rejuvenated antibody response, with titers rebounding to levels intermediate between those of young and old untreated mice (161.3±44.8, p=0.03 vs. young).
Further Research ... and Development
It remains to be demonstrated that this apparent rejuvenation effect extends to a gold-standard test of greater survival from infectious disease in treated animals. If that can be convincingly shown, then discovering the reasons for the remaining limits on B-cell function in animals treated with B-cell depletion will be important to further the progress to this research, and to using the proof-of-principle that the Israel-Technion team appears to have provided to develop intervention protocols suitable not only for animal testing, but for translation into rejuvenation therapies for aging humans.
Another question is how these new findings integrate with prior research on the aging haematopoietic system. Previous work has shown the role of the deranged signaling environment of the biologically aged organism in repressing non-immune functions of the haematopoietic stem cell niche, and the rejuvenation of the HSC niche by a youthful systemic environment. Other research has highlighted the role of intrinsic defects in the degenerative aging process of HSCs,(5-7) and the ability of the autophagy-enhancing (and, in mice, life-extending) drug rapamycin to partially restore immune function in aging mice.(8) The combination of this research on the systemic, cell-intrinsic, and population influences on degenerative aging of B-lymphocyte functions, and the many interventions already shown to partially rejuvenate aspects of it, gives significant grounds for optimism that interventions can be developed to effect a protocol -- or combination of protocols -- to effect the thoroughgoing rejuvenation of haematopoietic aging, and on timescales that many in the field might until recently have thought unrealistic.
For some time, SENS Foundation's research investments in immune rejuvenation have been centered on T-cell function, dedicated to the complementary strategies of engineering youthful thymic epithelium to restore youthful production of naïve T-cells, and ablation of anergic T-cells to open up the immunologic 'space' required for their expansion.(9,10) The finding that depletion of long-resident B-lymphocytes leads to a partial rejuvenation of B-cell precursor content, lymphopoieisis, and antibody production is an unexpected and striking parallel to the expected effects of ablating anergic T-cells. This new research strongly confirms the expectation that a comprehensive strategy of rejuvenation biotechnologies will need to include both arms of the adaptive immune system, and strongly suggests key features of the tools that will be needed to do so.
References
1: High KP, D'Aquila RT, Fuldner RA, Gerding DN, Halter JB, Haynes L, Hazzard WR, Jackson LA, Janoff E, Levin MJ, Nayfield SG, Nichol KL, Prabhudas M, Talbot HK, Clayton CP, Henderson R, Scott CM, Tarver ED, Woolard NF, Schmader KE. Workshop on immunizations in older adults: identifying future research agendas. J Am Geriatr Soc. 2010 Apr;58(4):765-76. PubMed PMID: 20398161.
2: Castle SC. Clinical relevance of age-related immune dysfunction. Clin Infect Dis. 2000 Aug;31(2):578-85. Epub 2000 Sep 14. Review. PubMed PMID: 10987724.
3: Pawelec G, Larbi A. Immunity and ageing in man: Annual Review 2006/2007. Exp Gerontol. 2008 Jan;43(1):34-8. Epub 2007 Oct 1. Review. PubMed PMID: 17977683.
4: Keren Z, Naor S, Nussbaum S, Golan K, Itkin T, Sasaki Y, Schmidt-Supprian M, Lapidot T, Melamed D. B cell depletion reactivates B lymphopoiesis in the BM and rejuvenates the B lineage in aging. Blood. 2011 Jan 12. [Epub ahead of print] PubMed PMID: 21228330.
5: Guerrettaz LM, Johnson SA, Cambier JC. Acquired hematopoietic stem cell defects determine B-cell repertoire changes associated with aging. Proc Natl Acad Sci U S A. 2008 Aug 19;105(33):11898-902. Epub 2008 Aug 12. PubMed PMID: 18697924; PubMed Central PMCID: PMC2515225.
6: Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005 Jun 28;102(26):9194-9. Epub 2005 Jun 20. PubMed PMID: 15967997; PubMed Central PMCID: PMC1153718.
7: Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006 Sep 28;443(7110):421-6. Epub 2006 Sep 6. PubMed PMID: 16957735.
8: Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009 Nov 24;2(98):ra75. PubMed PMID: 19934433.
9: Rebo J, Causey K, Zealley B, Webb T, Hamalainen M, Cook B, Schloendorn J. Whole-animal senescent cytotoxic T cell removal using antibodies linked to magnetic nanoparticles. Rejuvenation Res. 2010 Apr-Jun;13(2-3):298-300. PubMed PMID: 20426617.
10: Nikolich-Zugich Lab. Rejuvenation of the aging t-cell pool by rebalancing T-cell repertoire. DG Cohort #1 Final Report. 2010 Nov 23.
Posted 03 January 2011 - 11:40 PM
Milestones achieved in 2010: The creation of an internal A2E synthesis capability enabled efficient target enzyme evaluation. Our screening and development of several protein purification protocols resulted in the isolation of an active, A2E-degrading enzyme that is engineered to have features that make it interesting from a clinical perspective.
One of our priorities at the SENS Foundation Research Center has been to overcome the limitations of our ability to test potential A2E-degrading enzymes caused by a restricted supply of the target molecule (A2E). We have now successfully synthesized A2E, purifying it by flash chromatography and HPLC, and confirming it to be identical to that produced in Janet Sparrow's laboratory through different analytical tests. Our ability to produce this material "in house" is a key element in the expansion of our screening and evaluation of both existing and newly-purified A2E-degrading enzymes.
In collaboration with students at the State University of New York at Plattsburgh, we have also made significant progress in dramatically improving the A2E-degrading capacity of one of our enzymes after fusing it with a second enzyme. While the former catabolizes the target molecule, destroying its toxic activity, the latter produces a metabolite that accelerates the degradation activity up to a hundredfold.
The year ahead: We will express and purify additional candidate enzymes for both macular degeneration and atherosclerosis projects, exploiting new biochemical strategie; establish activity for these enzymes in vitro; perform lysosomal-uptake studies in RPE cells for macular degeneration and in macrophages for 7-ketocholesterol; and perform initial toxicity studies.
Posted 17 February 2011 - 10:24 PM
The often-mooted question of whether "aging itself" is or is not a "disease" has long been mooted in biogerontological circles, with a long-held rhetorical preference for asserting that it is not, but rather, that it is a risk factor for the specific diseases of aging.(1) By contrast, the same fundamental semantic dispute was initially resolved in the opposite direction with regard to age-related cognitive decline and dementia, beginning in the early decades after Alois Alzheimer and Emil Kraepelin first identified the pathological basis of the Alzheimer's disease (AD) until the early 1970s. For most of the twentieth century, it was held that dementia occurring in younger people should be classified as a disease, whereas dementia should be expected and accepted when it occurred in people at more advanced ages, despite the knowledge that the lesions linked to Alzheimer's dementia accumulated throughout the course of "normal" aging in middle age and onward, and that the pathological basis of the disorder was the same in both cases.(2)
But beginning in the 1960s, a loose alliance led by social gerontologists but quickly coming to include biogerontologists, geriatricians, and patient advocacy groups successfully campaigned for a new understanding: that while some level of minor cognitive decline was indeed a "normal" and inevitable part of aging, the newly-rediscovered clinicopathological entity, "Alzheimer's disease," was exactly that: a disease, against which the full force of public and private biomedical research should be mobilized in the pursuit of a cure.(2)
The veneer of coherence to this division has been peeling away for some years now, with the identification of mild cognitive impairment (MCI) as a prodromal or "preclinical" stage of AD, with cerebrospinal, pathological, and neuroimaging evidence linking it clearly to the core pathology of the clinical disease itself. Indeed, it is now well-established that aggregated beta-amyloid protein (Aβ) and neurofibrillary tangles (NFT -- cytoplasmic inclusions composed of phosphorylated and abnormally-cleaved species of tau protein) accumulate progressively in the "normally" aging brain and precede the onset of dementia by decades.(5,6) Two recent publications (3,4) clearly refute the principle arguments in favor of this dichotomy, firmly rooting the basis of "normal" age-related cognitive decline in the same pathological lesions of the brain that drive the "dementia" of Alzheimer's.
In one report,(3) researchers at the Rush Alzheimer's Disease Center used data from 354 older clergy from the prospective Religious Orders Study, who had undergone baseline and up to 13 annual followup rounds of medical and psychological testing which included sound composite measures of global cognition and of specific cognitive functions, and culminated in postmortem brain autopsy. In order to distinguish the long course of age-related cognitive decline from the widely-observed rapid phase of antemortem cognitive decline without falling into the petitio principii of using the clinical diagnostic criteria for dementia or Alzheimer's disease, the investigators tested a series of statistical models that each fit the observed rates of cognitive decline with an hypothesized acceleration in the last n months of life, and selected the best fit model as the analysis with the highest log likelihood value. The inflection point was thereby determined to occur at ~52 mo antemortem.(3)
Into this model they fit in the interactions of a pathologic index for lesions related to age-related cognitive decline and dementia: density of NFT, the lesion most strongly associated with the level of cognition in AD; Lewy bodies, the characteristic marker of Lewy body dementia (DLB); and gross and microscopic cerebral infarcts occurring at least 6 mo anemortem. as indicators of stroke and transient ischemic attack. With this analysis, they evaluated relationships between each pathological lesion, as well as a final model including all, with "normal" vs. "disease" related (accelerated terminal) decline.
The results (all emphasis mine):
higher tangle density was associated with more rapid age-related and disease-related decline in global cognition. ... [V]irtually no age-related change in global cognition occurred at low levels of tangles (25th percentile...) compared to substantial decline at high levels (75th percentile...). By contrast, much disease-related global cognitive decline occurred despite low levels of tangles, suggesting the involvement of other pathologic factors. ...
[B]oth gross ... and microscopic ... infarction were associated with a more than 2-fold increase in rate of age-related global cognitive decline. By contrast, neither gross nor microscopic infarction was associated with disease-related [global] cognitive decline. ... The presence of neocortical Lewy bodies ... was associated with an approximate doubling of disease-related decline relative to those without Lewy bodies ... and [with] a nearly significant effect on age-related decline. By contrast, nigral/limbic Lewy bodies [characteristic of the movement disorders of Parkinson's disease] ... were not associated with either age-related or disease-related decline in global cognition ...Higher tangle density was associated with more rapid age-related and disease-related decline in all cognitive systems ... By contrast, cerebral infarction ... and Lewy bodies ... had selective effects across time and cognitive systems. ... It is noteworthy that Lewy bodies were associated with decline in episodic memory, a defining characteristic of AD, and that all forms of pathology contributed to age-related decline in working memory, a change often attributed to normal aging. ...With all pathologic measures in the same model, tangles continued to be associated with age-related and disease-related decline in multiple cognitive systems; gross infarction was associated with age-related working memory decline; and neocortical Lewy bodies were associated with age-related perceptual speed decline and disease-related decline in episodic and semantic memory ...
Age-related cognitive decline was associated with neurofibrillary tangles, cerebral infarction, and Lewy bodies, and was not evident in the absence of these lesions. This indicates that the neurodegenerative lesions traditionally associated with dementia are principally responsible for the gradual age-related cognitive decline that precedes dementia and that AD and related disorders have a much greater impact on late-life cognitive functioning than previously recognized. ...
Gradual cognitive decline in old age has mainly been thought to reflect normative age-related developmental processes. In this cohort, however, there was no age-related cognitive decline absent postmortem evidence of neurodegenerative disease, and multiple pathologic lesions were associated with rate of age-related cognitive decline. These data challenge the concept of normative cognitive aging and suggest instead that neurodegenerative disease plays a role in virtually all late-life cognitive decline ...
The results also indicate that factors other than tangles and neocortical Lewy bodies are contributing to variability in disease-related [terminal] cognitive decline. This could include other pathologic features such as the TAR DNA-binding protein 43 [TDP-43]. In addition, neurodegeneration in the form of loss of neurons and synapses may be the most proximate cause of precipitous cognitive decline, leaving less variability to be accounted for by more distal contributors to neurodegeneration such as tangles and Lewy bodies.(3)
The second paper(4) gave context to this latter allusion: the fact that neuron loss is minimal in the "normally" aging brain, but is prevalent in the late stages of dementia. This relatively recent observation was greeted with surprise, because it stood in curious contrast to the much longer-established fact of extensive volumetric shrinkage of the brain across the course of "normal" cognitive aging, which had previously been assumed to imply a lifelong process of neuronal loss. Once it became clear that little such loss occurred, this fact was often invoked as evidence of a clear distinction between the age-related changes occurring in "normal" cognitive aging and the pathological processes contributing to dementia. But it left the exact structural basis of age-related brain shrinkage largely unexplained.
The new study(4) shows that this long process of brain shrinkage during "normal" aging is largely the result, not of neuronal loss, but of premorbid neuronal atrophy:
This paper reviews recent evidence from magnetic resonance imaging (MRI) studies about age-related changes in the brain. The main conclusions are that
(1) the brain shrinks in volume and the ventricular system expands in healthy aging. However, the pattern of changes is highly heterogeneous, with the largest changes seen in the frontal and temporal cortex, and in the putamen, thalamus, and accumbens. With modern approaches to analysis of MRI data, changes in cortical thickness and subcortical volume can be tracked over periods as short as one year, with annual reductions of between 0.5% and 1.0% in most brain areas.
(2) The volumetric brain reductions in healthy aging are likely only to a minor extent related to neuronal loss. Rather, shrinkage of neurons, reductions of synaptic spines, and lower numbers of synapses probably account for the reductions in grey matter. In addition, the length of myelinated axons is greatly reduced, up to almost 50%.
(3) Reductions in specific cognitive abilities--for instance processing speed, executive functions, and episodic memory--are seen in healthy aging. Such reductions are to a substantial degree mediated by neuroanatomical changes, meaning that between 25% and 100% of the differences between young and old participants in selected cognitive functions can be explained by group differences in structural brain characteristics.(4)
Ultimately, whether we speak of aging as a biological process that can be separated from specific age-related diseases, or as the premorbid structural basis of age-related disease and frailty, or as itself the ultimate age-related disease, should be regarded as a conceptual convenience bordering on a literary conceit, of no ultimate clinical significance. The aging of the body is a process of accumulating cellular and molecular lesions that degrade the fidelity of the structural basis of normal homeostasis; from an heuristic point of view, it can and should be understood to be a pathological process with a biomedical solution. The strategy of rejuvenation biotechnology is to remove, repair, replace, or render harmless such lesions, restoring the structural integrity of the body to the original order seen in youth. In the process, we will restore health and vigor that emerges from youthful biological structures, eliminating age-related disease and disability even as we eliminate the damage that underlies it.
Referenes
1: de Grey AD. Resistance to debate on how to postpone ageing is delaying progress and costing lives. Open discussions in the biogerontology community would attract public interest and influence funding policy. EMBO Rep. 2005 Jul;6 Spec No:S49-53. PubMed PMID: 15995663; PubMed Central PMCID: PMC1369265.
2: Ballenger JF. Progress in the history of Alzheimer's disease: the importance of context. J Alzheimers Dis. 2006;9(3 Suppl):5-13. PubMed PMID: 17004361.
3: Wilson RS, Leurgans SE, Boyle PA, Schneider JA, Bennett DA. Neurodegenerative basis of age-related cognitive decline. Neurology. 2010 Sep 21;75(12):1070-8. Epub 2010 Sep 15. PubMed PMID: 20844243; PubMed Central PMCID: PMC2942064.
4: Fjell AM, Walhovd KB. Structural brain changes in aging: courses, causes and cognitive consequences. Rev Neurosci. 2010;21(3):187-221. Review. PubMed PMID: 20879692.
5:Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
6: Braskie MN, Klunder AD, Hayashi KM, Protas H, Kepe V, Miller KJ, Huang SC, Barrio JR, Ercoli LM, Siddarth P, Satyamurthy N, Liu J, Toga AW, Bookheimer SY, Small GW, Thompson PM. Plaque and tangle imaging and cognition in normal aging and Alzheimer's disease. Neurobiol Aging. 2010 Oct;31(10):1669-78. Epub 2008 Nov 11. PubMed PMID: 19004525; PubMed Central PMCID: PMC2891885.
Posted 22 March 2011 - 10:34 AM
I'm delighted to be able to share with you our research report, prepared for the first 10 months of 2010, by Tanya Jones (our Director of Research Operations), working with our researchers and my CSO Team. I thought it would be of interest to our supporters, and serve as a precursor to our 2010 Year End Report, which is currently under production as part of our finalizing our 2010 accounts. 2011 looks set to bring even greater progress in our mission, and we're busy designing the next iteration of our website, which will continue to improve our ability to deliver information about our extramural and intramural research activities.
Cheers,
Aubrey









0 members, 1 guests, 0 anonymous users


Community Forum Software by IP.Board
Licensed to: ImmInst.org




