SENS NEWS
 ImmInst
21 Mar 2011
ImmInst
21 Mar 2011
The early free radical theory of aging was based on Dr. Denham Harman's remarkable insight that much of the cellular and molecular damage of aging bore strong resemblance to damage he observed in organisms exposed to ionizing radiation.(1) In subsequent decades, the theory -- and its later refinement into the mitochondrial free radical theory of aging(2,3) -- has gained wide acceptance, even as the challenges against it have mounted and risen in sophistication. Despite the clear evidence that certain sources of reactive oxygen species (ROS*), and certain kinds of oxidative stress and damage, contribute to the degenerative aging process,(4,5) it has become equally clear that the early, and still widely-invoked, oversimplification that all free radicals of any sort, in any context, are deleterious, cannot be sustained.
Perhaps the most profound exception to the rule of the inherent toxicity of ROS can be laid at the feet of the engines of evolution. Over evolutionary time, natural selection has "learned" to harness these ubiquitous reactive small molecules for use in cellular signal transduction, playing initially-surprising but ultimately-comprehensible roles in such processes as insulin signaling and the adaptive response to resistance exercise.(6,24) A common mechanism of redox signaling is via chemical reactions of specific ROS (primarily singlet oxygen or hydrogen peroxide) with specific atoms (usually a cysteine residue) in transcription factors or other regulatory elements: the ensuing covalent modification of the protein alters its activity, providing a mechanism for the cell to respond dynamically to shifts in intracellular redox tone.

Figure 1. Bacterial examples of ROS regulation of transcription factors. Reproduced from (24).
Two recent studies have now provided especially salient examples of this novel form of the "oxygen paradox."
Antioxidants Impair Autophagy
David Rubinsztein's laboratory at the Cambridge Institute for Medical Research focuses on diseases of aggregate-prone proteins, and especially disorders caused by codon reiteration mutations, such as Huntington’s disease (HD). In the earlier study,(7) Dr. Ben Underwood and colleagues in Rubinsztein's lab followed up on earlier research documenting the role of ROS in mediating the upregulation of autophagy in response to starvation and a range of pharmacological agents, including the prominent case of rapamycin.
The importance of autophagy and oxidative stress to both pathology and potential therapies for neurodegeneration has led us to investigate their relationship further. A diverse range of stimuli that induce both ROS and autophagy have been described and autophagy induction by these agents is antagonized by antioxidants ... Here we show that not all autophagy inducers significantly increase ROS. However, many antioxidants inhibit both basal and induced autophagy ...When autophagy induction is mediated by an increase in ROS, increases in autophagy can be inhibited with ROS scavengers. We next tested whether the impairment of autophagy by antioxidants was limited to situations where the autophagy-inducing agent also significantly increased ROS. We tested the ability of a variety of thiol antioxidants (including those proposed for treatment of HD) to ameliorate the induction of autophagy by trehalose (which does not increase levels of ROS) in COS-7 cells.
We found that NAC N-acetylcysteine (NAC), cystamine (a drug proposed for use in HD ... which is also metabolized to the antioxidant L-cysteine (19)) and glutathione were all able to significantly ameliorate the induction of autophagy by trehalose in a dose-dependent fashion, as measured by levels of the autophagy marker, microtubule-associated protein 1 light chain 3 II (LC3-II) ... NAC [also] impaired the increase in LC3-II associated with rapamycin treatment and also decreased LC3-II compared with the basal state. We confirmed the effect of NAC on basal and inducible autophagy in human primary cortical neurons ... and in HeLa cells (Fig. 2F). Pre-treatment with cystamine had similar effects on basal autophagy ...
[W]e examined [several antioxidants'] effect on the mTOR [mammalian Target of Rapamycin] pathway, a classical negative regulator of autophagy, by looking for changes in the phosphorylation status of an mTOR kinase substrate 4E binding protein 1 (4E-BP1) and a protein dependent on mTOR substrate kinase activity, ribosomal protein S6 (S6). Vitamin E enhanced the activity of mTOR but, surprisingly, NAC appeared to be inhibiting mTOR activity—an effect that would be expected to increase, rather than decrease, autophagy. ...
Starvation-induced autophagy is associated with increased levels of ROS and has been shown to be mediated by the activation of c-Jun N-terminal protein kinase 1 (JNK1) ... NAC is able to inhibit both ROS accumulation and autophagy in starved cells. ... NAC and glutathione inhibited the activation of JNK and decreased the phosphorylation of Bcl-2. ...
In order to investigate the physiological relevance of our findings, we examined the effect of NAC on starvation-induced autophagy in mice. ... As expected, starvation strongly increased hepatic LC3-II levels, but this increase was significantly smaller in mice that had been pre-treated with NAC. ...
By blocking autophagy, antioxidant drugs can increase the levels of aggregate-prone proteins associated with neurodegenerative disease. In fly and zebrafish models of Huntington's disease, antioxidants exacerbate the disease phenotype and abrogate the rescue seen with autophagy-inducing agents.Thus, the potential benefits in neurodegenerative diseases of some classes of antioxidants may be compromised by their autophagy-blocking properties.(7)
From an evolutionary point of view, one can see a highly-intuitive hypothesis to explain of this phenomenon. When damaged biomolecules begin to accumulate in the cell, it will often lead to oxidative stress downstream; equally, conditions of high oxidative stress will predictably lead to an increase in the production of such damaged cellular constituents. This could create selective pressure favoring the retention of redox-sensitive protein sites on regulatory elements in the degradative machinery, allowing for upregulation of the proteasomal or (in this case) lysosomal machinery to clear the offending aggregates.
ROS-Sensitive Regulation of Stem Cell Proliferative Homeostasis
Perhaps more surprising is a more recent report from Heinrich Jasper's lab at the University of Rochester, showing that the antioxidant Cap'n'collar transcription factor Nrf2 -- which suppresses intracellular oxidative stress by inducing genes involved in the production of the cellular antioxidants glutathione and the thoredoxins, along with peroxiredoxin enzymes -- is constitutively active in Drosophila intestinal stem cells.(8) The high level of CncC (Drosophila Cap'n'collar group, including Nrf2) activity in intestinal stem cells substantially blunts the paraquat-induced rise in oxidative stress, while RNA interference (RNAi)-mediated repression of CncC translation leads to a rise in basal oxidative stress and elevated stem cell proliferation.
The role of ROS in regulating stem cell proliferation was also confirmed by other means. Thus, knockdown components of complex I of the mitochondrial electron transport chain via RNAi leads to a rise in intracellular ROS concentration, with a concomitant rise in stem cell proliferation. Contrariwise, overexpression the genes encoding the rate-limiting enzyme for glutathione biosynthesis, or of those encoding a thioredoxin peroxidase whose expression can be stimulated by CncC overexpression, simultaneously lowers ROS concentration and proliferation rate in these stem cells. Most dramatically, overexpression of these same genes also delays the onset of the age-associated increase in gut stem cell mitosis observed in wild-type flies.(8)
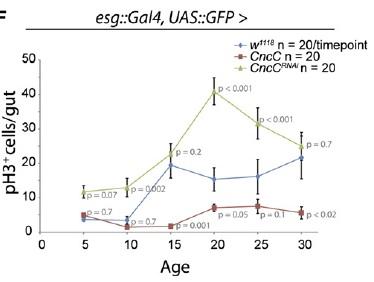
Figure 2. Elevating CncC Activity (and Nrf2) Delays Age-Related Rise in Uncontrolled Stem Cell Proliferation. Reproduced from (8).
Thus, Nrf2-induced suppression of ROS in these cells was shown to enforce quiescence. The counterintuitive corollary of this finding is that in order to respond to inflammation and oxidative stress with stem cell proliferation to ensure tissue renewal, the "protective" antioxidant-induction activity of Nrf2 must be repressed. Accordingly, induced coexpression of Nrf2/CncC was shown to inhibit the defensive proliferative response induced by a range of stimuli, including oxidative stress (paraquat), mitogenic signaling through the insulin receptor, expression of the stress-response protein JNK (which is normally activated by stressors such as inflammatory cytokines, heat or osmotic shock, ultraviolet irradiation, and bacterial enterotoxins), by exposure to bacterial lipopolysaccharide, or by hyperplasia induced by Notch-targeting RNAi.(8) The negative regulation required to rein in Nrf2 and allow stem cells to proliferate under stress conditions was shown to be provided by Keap1, which sequesters Nrf2 in the cytosol and ultimately ubiquitinates it for subsequent proteolytic degradation.(8)
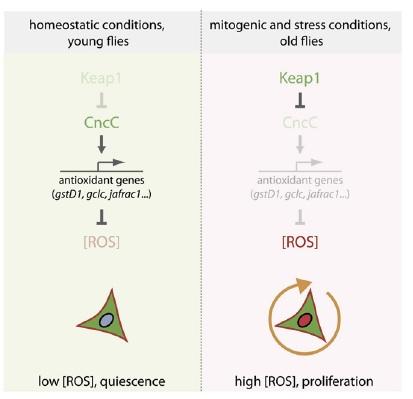
Figure 3. Nrf2, ROS, and the Regulation of Stem Cell Proliferation. Reproduced from (8).
These findings shed light on an earlier report from the same group:
In aging flies, the intestinal epithelium degenerates due to over-proliferation of intestinal stem cells (ISCs) and mis-differentiation of ISC daughter cells, resulting in intestinal dysplasia. Here we show that conditions that impair tissue renewal [ectopic activation of Notch, or overexpression of JNK or insulin signaling] lead to lifespan shortening, whereas genetic manipulations that improve proliferative homeostasis extend lifespan. These include reduced Insulin/IGF or Jun-N-terminal Kinase (JNK) signaling activities, as well as over-expression of stress-protective genes [the heat-shock protein Hsp68 or the peroxiredoxin Jafrac1] in somatic stem cell lineages. Interestingly, proliferative activity in aging intestinal epithelia correlates with longevity over a range of genotypes, with maximal lifespan when intestinal proliferation is reduced but not completely inhibited [my emphasis]. Our results highlight the importance of the balance between regenerative processes and strategies to prevent hyperproliferative disorders and demonstrate that promoting proliferative homeostasis in aging metazoans is a viable strategy to extend lifespan.(9)
Figure 4. Mild Repression of Stem Cell Proliferation Extends Lifespan in Drosophila. Reproduced from (9).
Implications for Intervention in Aging
Long prior to these recent findings, earlier research not discussed by these authors had shown that life-extending Calorie restriction (CR) in rodents exhibits this same property, placing cell replication under a more "thrifty" regulatory regime and thereby maintaining tisue-renewal capacity with age (16-19) and contributing to the CR animals' cancer resistance (20-23) and extended lifespan.(21) Established mediators of this more aggressive control of cell proliferation include reduced signaling through the insulin, insulin-like growth factor, and mammalian target of rapamycin (mTOR) signaling pathways. More recently, and apparently independently of all three lines of inquiry, still another group has reported that much of the cancer-protective effect of CR in mice is dependent on upregulation of Nrf2 activity.(10) Taken together, these findings intimate the possibility that the same Nrf2-dependent control of stem cell proliferation, dysplasia, and lifespan observed in Drosophila(8,9) may extend to the well-established rodent CR model of retarded aging.(10,16-23)
Interestingly, several dietary phytochemicals thought to have chemopreventive activity have also been shown to increase Nrf2 activity in vivo in at least some tissues after oral administration.(11-15) It is generally assumed that the mechanistic basis for the apparent role of Nrf2 induction in a cancer-preventive action of such agents and of CR is through the quenching of ROS and the detoxification of carcinogenic and mutagenic compounds.(10-15) This new research(8) suggests Nrf2-mediated inhibition of stem cell proliferation as an additional or alternative mechanism.
As Underwood et al also highlight for their own research into autophagy,(7) the new revelations about Nrf2 in regulation of stem cell proliferation(8-10) suggests the possibility that antioxidant supplementation may impede the protective effects of these interventions. Such pleiotropic effects could hypothetically have contributed to the multiple failures of antioxidant therapies to extend lifespan in experimental animals, or to reduce the incidence of cardiovascular disease and cancer in large clinical trials in humans. More speculatively, it may also raise some concerns about efforts to restore stem cell mobilization in aging tissues by elevating Notch signaling. And further complicating the interpretation of these findings or their use as the basis for interventions in age-related disease, mutations leading to constitutive activation of Nrf2 have been associated with several kinds of cancer -- most prominently of the lung -- in humans.(25)
As in previous cases we have highlighted, paradoxical findings and dilemmas such as these challenge the wisdom of efforts to retard the degenerative aging process by manipulating metabolic regulatory machinery and its dynamic intermediates. Homeostasis relies upon the ability of the organism to sense its internal and external environment, and respond to it adaptively; our best efforts to improve on the homeostatic regulatory machinery, or to selectively alter the dynamic environment itself, will inevitably tend to founder on the rocks of our ignorance of metabolism, and sink in the swirling seas of its complexity. The "engineering" heuristic of intervention in the degenerative aging process offers a means to traverse these perilous waters by figuratively flying above them, leaving the regulation of homeostasis to the finely-tuned machinery that has already been shaped by the complex calculus of deep evolutionary time -- machinery which has kept ourselves, our species, and our evolutionary ancestors alive for all of our lives. Rather, rejuvenation biotechnology will maintain youthful health and vigor through the direct repair, removal, replacement, and rendering-harmless of the damage itself instead of second-guessing its regulators.
Note
* I will here adhere to this conventional terminological convenience, with the understanding that the species under discussion include some molecules and ions that do not meet the literal meaning of the term, such as reactive nitrogen species and reduced transition metal ions.
References
1: Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956 Jul;11(3):298-300. PubMed PMID: 13332224.
2: Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972 Apr;20(4):145-7. PubMed PMID: 5016631.
3: de Grey ADNJ. The mitochondrial free radical theory of aging. Austin, TX: Landes Bioscience, 1999, 212pp, hardcover (ISBN 1-57059-564-X).
4: Pamplona R, Barja G. Highly resistant macromolecular components and low rate of generation of endogenous damage: two key traits of longevity. Ageing Res Rev. 2007 Oct;6(3):189-210. Epub 2007 Jul 13. Review. PMID: 17702671 [PubMed - indexed for MEDLINE]
5: Pamplona R. Mitochondrial DNA Damage and Animal Longevity: Insights from Comparative Studies. J Aging Res. 2011, Article ID 807108, 9 pages. doi:10.4061/2011/807108
6: Rudolph TK, Freeman BA. Transduction of redox signaling by electrophile-protein reactions. Sci Signal. 2009 Sep 29;2(90):re7. Review. PubMed PMID: 19797270.
7: Underwood BR, Imarisio S, Fleming A, Rose C, Krishna G, Heard P, Quick M, Korolchuk VI, Renna M, Sarkar S, García-Arencibia M, O'Kane CJ, Murphy MP, Rubinsztein DC. Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum Mol Genet. 2010 Sep 1;19(17):3413-29. Epub 2010 Jun 21. PubMed PMID: 20566712; PubMed Central PMCID: PMC2916709.
8: Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011 Feb 4;8(2):188-99. PubMed PMID: 21295275; PubMed Central PMCID: PMC3035938</p>Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011 Feb 4;8(2):188-99. PubMed PMID: 21295275; PubMed Central PMCID: PMC3035938
9. Biteau B, Karpac J, Supoyo S, Degennaro M, Lehmann R, Jasper H. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 2010 Oct 14;6(10):e1001159. PubMed PMID: 20976250; PubMed Central PMCID: PMC2954830.
10: Martín-Montalvo A, Villalba JM, Navas P, de Cabo R. NRF2, cancer and calorie restriction. Oncogene. 2011 Feb 3;30(5):505-20. Epub 2010 Nov 8. PubMed PMID: 21057541.
11: McWalter GK, Higgins LG, McLellan LI, Henderson CJ, Song L, Thornalley PJ, Itoh K, Yamamoto M, Hayes JD. Transcription factor Nrf2 is essential for induction of NAD(P)H:quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J Nutr. 2004 Dec;134(12 Suppl):3499S-3506S. PubMed PMID: 15570060.
12: Shen G, Xu C, Hu R, Jain MR, Gopalkrishnan A, Nair S, Huang MT, Chan JY, Kong AN. Modulation of nuclear factor E2-related factor 2-mediated gene expression in mice liver and small intestine by cancer chemopreventive agent curcumin. Mol Cancer Ther. 2006 Jan;5(1):39-51. PubMed PMID: 16432161.
13: Yuan JH, Li YQ, Yang XY. Inhibition of epigallocatechin gallate on orthotopic colon cancer by upregulating the Nrf2-UGT1A signal pathway in nude mice. Pharmacology. 2007;80(4):269-78. Epub 2007 Jul 26. PubMed PMID: 17657175.
14: Yuan JH, Li YQ, Yang XY. Protective effects of epigallocatechin gallate on colon preneoplastic lesions induced by 2-amino-3-methylimidazo[4,5-f ] quinoline in mice. Mol Med. 2008 Sep-Oct;14(9-10):590-8. PubMed PMID: 18596869; PubMed Central PMCID: PMC2442020.
15: Balstad TR, Carlsen H, Myhrstad MC, Kolberg M, Reiersen H, Gilen L, Ebihara K, Paur I, Blomhoff R. Coffee, broccoli and spices are strong inducers of electrophile response element-dependent transcription in vitro and in vivo - Studies in electrophile response element transgenic mice. Mol Nutr Food Res. 2011 Feb;55(2):185-97. doi: 10.1002/mnfr.201000204. Epub 2010 Sep 8. PubMed PMID: 20827676.
16: Chou MW, Shaddock JG, Kong J, Hart RW, Casciano DA. Effect of dietary restriction on partial hepatectomy-induced liver regeneration of aged F344 rats. Cancer Lett. 1995 May 8;91(2):191-7. PubMed PMID: 7767909.
17:Wolf NS, Penn PE, Jiang D, Fei RG, Pendergrass WR. Caloric restriction: conservation of in vivo cellular replicative capacity accompanies life-span extension in mice. Exp Cell Res. 1995 Apr;217(2):317-23. PubMed PMID: 7698231.
18: Holcomb VB, Keck VA, Barrett JC, Hong J, Libutti SK, Nunez NP. Obesity impairs wound healing in ovariectomized female mice. In Vivo. 2009 Jul-Aug;23(4):515-8. PubMed PMID: 19567384.
19: Reed MJ, Penn PE, Li Y, Birnbaum R, Vernon RB, Johnson TS, Pendergrass WR, Sage EH, Abrass IB, Wolf NS. Enhanced cell proliferation and biosynthesis mediate improved wound repair in refed, caloric-restricted mice. Mech Ageing Dev. 1996 Jul 31;89(1):21-43. PubMed PMID: 8819104.
20: Bursch W, Grasl-Kraupp B, Wastl U, Hufnagl K, Chabicovsky M, Taper H, Schulte-Hermann R. Role of apoptosis for mouse liver growth regulation and tumor promotion: comparative analysis of mice with high (C3H/He) and low (C57Bl/6J) cancer susceptibility. Toxicol Lett. 2004 Apr 1;149(1-3):25-35. Review. PubMed PMID: 15093245.
21: Koizumi A, Wada Y, Tuskada M, Kayo T, Naruse M, Horiuchi K, Mogi T, Yoshioka M, Sasaki M, Miyamaura Y, Abe T, Ohtomo K, Walford RL. A tumor preventive effect of dietary restriction is antagonized by a high housing temperature through deprivation of torpor. Mech Ageing Dev. 1996 Nov 29;92(1):67-82. PubMed PMID: 9032756.
22: Koizumi A, Tsukada M, Hirano S, Kamiyama S, Masuda H, Suzuki KT. Energy restriction that inhibits cellular proliferation by torpor can decrease susceptibility to spontaneous and asbestos-induced lung tumors in A/J mice. Lab Invest. 1993 Jun;68(6):728-39. PubMed PMID: 8515658.
23: Jin YH, Koizumi A. Decreased cellular proliferation by energy restriction is recovered by increasing housing temperature in rats. Mech Ageing Dev. 1994 Jul;75(1):59-67. PubMed PMID: 9128754.
24: D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007 Oct;8(10):813-24. Review. PubMed PMID: 17848967.
25: Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009 Apr;34(4):176-88. Epub 2009 Mar 25. Review. PubMed PMID: 19321346.
View the full article
ImmInst
27 Mar 2011
Much of the distraction in the literature of biogerontology, and an even higher ratio of studies cited and promoted in the popular media and the dietary supplement industry, derives from methodologically-poor lifespan studies in mice (or occasionally rats). The pineal hormone melatonin,(1) the wine polyphenol resveratrol,(2) branched-chain amino acids,(3) the mitochondrially-targeted antioxidant plastoquinonyl decyltriphenyl phosphonium (SkQ1),(4,5) mice with a Fat-specific Insulin Receptor Knockout (FIRKO)(6), ribonucleic acid supplementation,(7) various vitamin mixtures,((8), misinterpretation of (11)'s honest reporting) the selective irreversible MAO-B inhibitor and Parkinson's disease treatment selegiline (L-deprenyl, Eldepryl)(9,10) ... a long series of instantiations of the Original Sin of biogerontology.
In these studies, an increase in mean or maximal lifespan is reported, relative to short-lived controls, and claimed to be informative about the universal, degenerative aging process and the prospects for extending healthy life in humans living in the developed world. And the claim typically persists for decades once widely-cited, despite the best efforts of serious investigators to critique weak methodologies and flawed interpretation (eg. (12,13)), or even robust demonstrations of a null effect in healthy animals (as the two independent demonstrations that resveratrol does not extend lifespan in nonobese, wild-type mice over a wide range of doses).
Extensive studies by careful investigators such as Dr. Stephen Spindler of UC Riverside, Dr. Richard Weindruch of the University of Wisconsin at Madison, and Dr. Richard Miller of the University of Michican have shown that careful husbandry of a healthy cohort of wild-type laboratory mice fed a nonobesogenic environment and maintained in specific pathogen-free laboratores will, on average, live to be ~900 days, with a cohort maximum lifespan (operationally defined as tenth-decile survivorship) of ~1100 d. But in report after report of 'life extension' in mice, either none of the animals -- controls or intervention animals -- reach even this threshold lifespan, or else only the intervention group does -- and this result is declared to be a significant advance in our understanding of the aging process and its modulation.
The aforementioned Dr. Spindler has done a great deal of important Calorie restriction (CR) research, including the study definitively establishing that CR continues to be effective in early seniority (19-20 mo old at initiation),(14) and is one of the few investigators to run genuinely rigorous mouse lifespan studies, thereby debunking multitudes of purported "anti-aging" dietary supplements (eg., (15)). Thus, the legitimate biogerontological community, and advocates of consumer protection, already owe a debt of gratitude to Spindler for his years of painstaking labor on our behalf in the laboratory.
Now, Spindler has written an extensive review of the many flaws that litter the litters in the literature, and proposed concrete methods to avoid these flaws and confounds:
Much of the literature describing the search for agents that increase the life span of rodents was found to suffer from confounds. One-hundred-six studies, absent 20 contradictory melatonin studies, of compounds or combinations of compounds were reviewed.
Only six studies reported both life span extension and food consumption data, thereby excluding the potential effects of caloric restriction. Six other studies reported life span extension without a change in body weight. However, weight can be an unreliable surrogate measure of caloric consumption. Twenty studies reported that food consumption or weight was unchanged, but it was unclear whether these data were anecdotal or systematic.
Twenty-nine reported extended life span likely due to induced caloric restriction. Thirty-six studies reported no effect on life span, and three a decrease. The remaining studies suffer from more serious confounds.
Though still widely cited, studies showing life span extension using short-lived or "enfeebled" rodents have not been shown to predict longevity effects in long-lived animals.
We suggest improvements in experimental design that will enhance the reliability of the rodent life span literature. First, animals should receive measured quantities of food and its consumption monitored, preferably daily, and reported. Weights should be measured regularly and reported. Second, a genetically heterogeneous, long-lived rodent should be utilized. Third, chemically defined diets should be used. Fourth, a positive control (e.g., a calorically restricted group) is highly desirable. Fifth, drug dosages should be chosen based on surrogate endpoints or accepted cross-species scaling factors. These procedures should improve the reliability of the scientific literature and accelerate the identification of longevity and health span-enhancing agents.(16)
I was surprised to see that the state of the literature is even worse than I had realized, as subtle flaws in execution and reporting that I'd've blithely passed over or accepted prove to be important to certainty about the meaning of the results, and bring into question several good-looking studies (although in most cases, reports with subtle flaws also have larger ones).
Happily, the full text of this comprehensive review is available online from the publisher -- a guide to designing, executing, and interpreting reports of rodent lifespan studies , by someone who knows from experience what is required to generate robust experimental results. There is much to gain from Spindler's years of experience, for investigators preparing to execute rodent longevity studies and for those seeking to understand the ensuing reports, for potential funders of such interventions, and for anyone wishing to become an informed reader of the literature on which progress toward an extension of healthy human lifespan may depend. This review should be considered required reading for those interested in advancing the science of aging rather than generating headlines or supplement sales.
References
1. Pierpaoli W, Dall'Ara A, Pedrinis E, Regelson W. The pineal control of aging. The effects of melatonin and pineal grafting on the survival of older mice. Ann N Y Acad Sci. 1991;621:291-313. PMID: 1859093 [PubMed - indexed for MEDLINE]
2: Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006 Nov 16;444(7117):337-42. Epub 2006 Nov 1. PMID: 17086191 [PubMed - indexed for MEDLINE]
3: D'Antona G, Ragni M, Cardile A, Tedesco L, Dossena M, Bruttini F, Caliaro F, Corsetti G, Bottinelli R, Carruba MO, Valerio A, Nisoli E. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab. 2010 Oct 6;12(4):362-72. PubMed PMID: 20889128.
4: Skulachev VP, Anisimov VN, Antonenko YN, Bakeeva LE, Chernyak BV, Erichev VP, Filenko OF, Kalinina NI, Kapelko VI, Kolosova NG, Kopnin BP, Korshunova GA, Lichinitser MR, Obukhova LA, Pasyukova EG, Pisarenko OI, Roginsky VA, Ruuge EK, Senin II, Severina II, Skulachev MV, Spivak IM, Tashlitsky VN, Tkachuk VA, Vyssokikh MY, Yaguzhinsky LS, Zorov DB. An attempt to prevent senescence: a mitochondrial approach. Biochim Biophys Acta. 2009 May;1787(5):437-61. Epub 2008 Dec 29. Review. PubMed PMID: 19159610.
5: Obukhova LA, Skulachev VP, Kolosova NG. Mitochondria-targeted antioxidant SkQ1 inhibits age-dependent involution of the thymus in normal and senescence-prone rats. Aging (Albany NY). 2009 Apr 22;1(4):389-401. PubMed PMID: 20195490; PubMed Central PMCID: PMC2830050.
6: Blüher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003 Jan 24;299(5606):572-4. PubMed PMID: 12543978.
7: Odens M. Prolongation of the life span in rats. J Am Geriatr Soc. 1973 Oct;21(10):450-1. PubMed PMID: 4729008.
8: Lemon JA, Boreham DR, Rollo CD. A complex dietary supplement extends longevity of mice. J Gerontol A Biol Sci Med Sci. 2005 Mar;60(3):275-9. PubMed PMID: 15860460.
9: Kitani K, Minami C, Isobe K, Maehara K, Kanai S, Ivy GO, Carrillo MC. Why (--)deprenyl prolongs survivals of experimental animals: increase of anti-oxidant enzymes in brain and other body tissues as well as mobilization of various humoral factors may lead to systemic anti-aging effects. Mech Ageing Dev. 2002 Apr 30;123(8):1087-100. Review. PMID: 12044958 [PubMed - indexed for MEDLINE]
10: Kitani K, Kanai S, Ivy GO, Carrillo MC. Assessing the effects of deprenyl on longevity and antioxidant defenses in different animal models. Ann N Y Acad Sci. 1998 Nov 20;854:291-306. Review. PMID: 9928438 [PubMed - indexed for MEDLINE]
11: Kokkonen GC, Barrows CH: The effect of dietary vitamin, protein and intake levels on the life span of mice of different ages. AGE.1985 Jan;8(1): 13-17.
12: Masoro EJ. A forum for commentaries on recent publications. FIRKO mouse report: important new model--but questionable interpretation. J Gerontol A Biol Sci Med Sci. 2003 Oct;58(10):B871-2. PubMed PMID: 14570851.
13: Anon. The stuff on which quackery thrives? Nutr Rev. 1974 Oct;32(10):316-7. PubMed PMID: 4416514.
14: Dhahbi JM, Kim HJ, Mote PL, Beaver RJ, Spindler SR. Temporal linkage between the phenotypic and genomic responses to caloric restriction. Proc Natl Acad Sci U S A. 2004 Apr 13;101(15):5524-9. Epub 2004 Mar 25. PubMed PMID: 15044709; PubMed Central PMCID: PMC397416.
15: Spindler SR, Mote PL. Screening candidate longevity therapeutics using gene-expression arrays. Gerontology. 2007;53(5):306-21. Epub 2007 Jun 15. Review. PubMed PMID: 17570924.
16: Spindler SR. Review of the literature and suggestions for the design of rodent survival studies for the identification of compounds that increase health and life span. Age (Dordr). 2011 Mar 22. [Epub ahead of print]. PMID: 21424790
View the full article
ImmInst
05 Apr 2011
It has been an exciting period ever since Dr. Doris Taylor of the University of Minnesota's Center for Cardiovascular repair outlined her results prior to publication in 2008 at the Foundation's Understanding Aging: Biomedical and Bioengineering Apporoaches conference at UCLA.
When the results were published, the popular press joined the field of rejuvenation biotechnology in hailing the major preclinical advance: the tissue engineering of a live, beating rat heart, generated using a decellularized myocardium as a scaffold, onto which cardiac stem cells were seeded.((1); see video of recellularized myocardial construct with tracer illustrating region of motion). These results were quickly expanded by Taylor's group as well as by independent investigators, who within two years were not only reporting similar results with reseeded decellularized lungs(2,3) and liver,(4) but the transplantation and in vivo functionality (albeit for brief periods) of these constructs. And the most recent advance came at the end of last year, with the announcement from Shay Soker, Anthony Atala, and colleagues at the Wake Forest Institute for Regenerative Medicine that
Livers from different species [mice, rats, ferrets, rabbits, and pigs] were perfused with detergent to selectively remove the cellular components of the tissue while preserving the extracellular matrix components and the intact vascular network. The decellularized vascular network was able to withstand fluid flow that entered through a central inlet vessel, branched into an extensive capillary bed, and coalesced into a single outlet vessel. The vascular network was used to reseed the scaffolds with human fetal liver and endothelial cells. These cells engrafted in their putative native locations within the decellularized organ and displayed typical endothelial, hepatic, and biliary epithelial markers, thus creating a liver-like tissue in vitro.[our emphasis](5)
Dr. Taylor had let it be known to us that she was pursuing work with human research, using decellularized tissue donations for the biological scaffolds reseeded with human stem cells -- but we were not aware of just how quickly she was making progress.
According to press reports in the Daily Mail and two stories in The Australian, "‘The hearts are growing, and we hope they will show signs of beating within the next weeks."
We emphasize, again, that these are press reports. Both sources assert that Dr. Taylor made the announcement at the American College of Cardiology’s 60th Annual Scientific Session and Innovation in Intervention (i2 Summit 2011),yet neither their meeting highlights nor the online abstract viewer appear to provide any information on this pending step forward.
With that caveat, and apologies for the conventions of the sources:
Live human heart grown in lab using stem cells in potential transplant breakthrough
By David DerbyshireThe scientists stripped the cells from the dead hearts with a powerful detergent, leaving ‘ghost heart’ scaffolds made from the protein collagen.
The ghost hearts were then injected with millions of stem cells, which had been extracted from patients and supplied with nutrients.
The stem cells ‘recognised’ the collagen heart structure and began to turn into heart muscle cells.
The hearts have yet to start beating – but if they do, they could be strong enough to pump blood.
However, the race to create a working heart faces many obstacles.
One of the biggest is getting enough oxygen to the organ through a complex network of blood vessels. Scientists also need to ensure the heart cells beat in time.
Dr Taylor told the Sunday Times: ‘We are a long way off creating a heart for transplant, but we think we’ve opened a door to building any organ for human transplant.’
Human hearts created in the lab have scientists excited
"The hearts are growing and we hope they will show signs of beating within the next week," said Doris Taylor, a specialist in regenerative medicine at the University of Minnesota. "There are many hurdles to overcome to generate a fully functional heart, but the hope is that it may one day be possible to grow entire organs for transplant." ...National Heart Foundation chief medical adviser James Tatoulis said the results were "an incredibly exciting breakthrough" ...
Jonathan Leake
“There are many hurdles to overcome to generate a fully functional heart, but the hope is that it may one day be possible to grow entire organs for transplant.” [said Dr. Taylor] ...
Dr Taylor points out that there is no shortage of pigs from which to extract hearts if no human cadavers are available. Once such a heart has been stripped of pig cells and reseeded with human stem cells taken from a patient needing a new heart, there should be few rejections.“We are a long way off creating a heart suitable for transplant, but the potential is clearly there," she said. ...
A key question for regenerative medicine researchers is how to make sure stem cells turn into the right thing - so they produce cardiac cells in the heart or liver cells in the liver.
Dr Taylor believes natural scaffolds help achieve this, partly because the stem cells recognise their shape. It may also be because they are each impregnated with chemicals specific to the organ from which they were derived. ...
“My ultimate goal is that one day we will be able to take a heart, probably from a pig, remove the cells and then replace them with cells grown from the patient's own body.“Then we would build a heart to match the patient and transplant it into them. That's the dream.”
Indeed it shall be, if these press reports are accurate, and if the resulting engineered myocardia prove even transiently viable: a key milestone in progress toward a comprehensive panel of rejuvenation biotechnologies.
Reference
1: Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, Taylor DA. Perfusion-decellularized matrix: using nature's platform to engineer a bioartificial heart. Nat Med. 2008 Feb;14(2):213-21. Epub 2008 Jan 13. PubMed PMID: 18193059.
2:. Petersen TH, Calle EA, Zhao L, Lee EJ, Gui L, Raredon MB, Gavrilov K, Yi T, Zhuang ZW, Breuer C, Herzog E, Niklason LE. Tissue-Engineered Lungs for in Vivo Implantation. . Science. 2010 Jun 28. [Epub ahead of print] PubMed PMID: 20576850.
3: Ott HC, Clippinger B, Conrad C, Schuetz C, Pomerantseva I, Ikonomou L, Kotton D, Vacanti JP. Regeneration and orthotopic transplantation of a bioartificial lung. Nat Med. 2010 Aug;16(8):927-33. Epub 2010 Jul 13. PubMed PMID: 20628374.
4: Uygun BE, Soto-Gutierrez A, Yagi H, Izamis ML, Guzzardi MA, Shulman C, Milwid J, Kobayashi N, Tilles A, Berthiaume F, Hertl M, Nahmias Y, Yarmush ML, Uygun K. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat Med. 2010 Jul;16(7):814-20. Epub 2010 Jun 13. PubMed PMID: 20543851; PubMed Central PMCID: PMC2930603.
5: Baptista PM, Siddiqui MM, Lozier G, Rodriguez SR, Atala A, Soker S. The use of whole organ decellularization for the generation of a vascularized liver organoid. Hepatology. 2010 Nov 12. [Epub ahead of print] PubMed PMID: 21225647.
View the full article
ImmInst
10 Apr 2011
Age-related accumulation of mutations in mitochondrial DNA (mtDNA) is widely suspected to play an important role in the degenerative aging process, albeit that controversy remains as to the mechanism(s) linking the two. Large deletions in mtDNA seem an especially likely culprit ...
A number of credible proposals have been advanced for rejuvenation biotechnology to restore youthful mitochondrial function in [cells homoplasmic for mitochondria bearing such deletions], reverting their abnormal metabolism and allowing them to resume participation in normal tissue function. The lead candidate approach, first proposed by SENS Foundation Chief Scientific Officer de Grey,(1) is the placement of functioning "backup copies" of the protein-coding mtDNA genes in the cell nucleus ("allotopic expression" (AE)). There has been substantial progress in this area since then,(eg (5-9)), and in recent years SENS Foundation has prioritized funding of AE research ...
But other potential routes to mitochondrial rejuvenation do exist and should also be developed, including the wholesale intraorganellar replacement of mtDNA using "protofection" (2) and the delivery of allotopic RNA to the organelle. The latter possibility was highligted by work [by Dr. Samit Adhya, of the Division of Molecular and Human Genetics at the Indian Institute of Chemical Biology] targeting tRNA human cell mitochondria with the transgenic use of a transfer RNA import complex [RIC] adapted from the parasitic protozoon Leishmania tropica.(3)
In that work, Dr. Adhya had demonstrated that the Leishmania RIC was efficiently taken up by human cells, where it was targeted to mitochondria and rescued oxidative phosphorylation (OXPHOS) in human cells haraboring the same tRNA mutations responsible for the inherited mitochondriopathy Myoclonic epilepsy with Ragged Red Fibers (MERRF).(3) In later work, his lab showed that the RIC could be used to induce the import of antisense oligonucleotides, leading to reduced expression of target mitochondrial mRNA.(4) And in a yet-unpublished preliminary proof-of-principle presented at the third Strategies for Engineered Negligible Senescence (SENS) conference in 2007,(9) Adhya demonstrated that the RIC-based system would work in vivo. Inherited mutations in ND1 subunit of mitochondrial complex I are responsible for the mitochondriopathy mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MERRF). As shown in dramatic video, injection of a RIC-based construct loaded with antisense RNA for ND1 into the rear left paws of wild-type rats lead to muscular degeneration, beginning with swelling and inflammation, and proceeding to severe slackening of the limb muscles, prominent movement impairments, and extensive myocyte necrosis -- a similar phenotype to the original human mitochondrial disease.(9)
Now, Adhya's group reports the next advance in this work, using a RIC complex to deliver large, functional memory RNA transcripts of mitochondrially-encoded electron transport chain (ETC) subunits into to cells lacking the genes those mRNA encode -- and to thereby rescue mitochondrial oxidative phosphorylation.
Kearns–Sayre Syndrome (KSS) is a mitochondriopathy characterized by ophthalmoparesis and ptosis (paralysis or weakness of the muscles that control eye and eyelid movement, respectively) as well as pigmentary retinopathy and abnormalities in cardiac conduction. To test the RIC system using large, mitochondrial mRNA, the investigators used a cybrid line derived from a KSS patient homoplasmic for a mtDNA deletion spanning from within the gene encoding Complex II to within that encoding Complex II. The cybrid line gene expression profile showed no expression of these genes, or of complex I, which the authors hypothesized might be due to destabilization of the transcript secondary to a processing defect brought on by the deletion.(10)
Researchers next generated a ribonucleoprotein complex of polycistronic RNA 1 that included mRNA for the genes along entire stretch of mtDNA from Complexes I through III, along with a mitochondrially-targeted tRNA import signal directly bound to the carrier complex via the tRNA receptors of R8, a functional recombinant subcomplex of RIC. Cells of the cybrid line were then transfected with the construct, termed pcRNA1-R8. Using several mitochondria-specific probes and subcellular fractionation, the researchers demonstrated a high degree of colocalization of fluorescently-labeled pcRNA1 mRNA with cybrid cells' mitochondria, with mitochondrial uptake reaching ~90% within 3 h. Adhya's group were were able to track the process pcRNA1-R8 binding, cellular uptake, cotransport with R8 to mitochondria, and the ultimate import into the mitochondrial matrix. Conversely, they demonstrated that there was no such localization in the case of pcRNA1 deprived of the R8 RIC subunit.(10)
Translation of the Mitochondrial Complex Proteins
The investigators next interrogated the cells for evidence of translation of the pcRNA1-R8-bound mRNA. Because the KSS mitochondrial deletion included the single mitochondrial tRNA for lysine, the cybrid lines suffered from global translational arrest. Exposing the cells to R8 alone restored translation of some mitochondrial genes, presumably by facilitating the import of tRNA from the cytosol, but not those for Complexes I-III. By contrast, pcRNA1-R8 was able to restore the wild-type synthesis pattern of all mitochondrial polypeptides; this was further confirmed for proteins specifically encoded by pcRNA1 using Western blotting, which additionally revealed their specific mitochonrial localization.(10)
The key question, however, is the ability of pcRNA1-R8 to restore the functioning of the electron transport chain revive oxidative phosphorylation in the cybrids.
Functional Mitochondria
ETC activity was confirmed in cybrid cells within 24 h of transfection with pcRNA1-R8 with the observation of functioning mitochondrial Complex IV activity; in turn, this implied the proper assembly of both the cybrids' missing subunits, and of the chain as a whole. More importantly, pcRNA1-R8 was also able to restore respiratory capacity, beginning with cellular oxygen uptake in the first 2-3 h of transfection, rising to ~93% of that of HepG2 liver cancer cells (which have wild-type mitochondria) within 24 h, and sustained for ~3d. Contrariwise, there was no stimulative (or inhibitory) effect of pcRNA1-R8 on HepG2 respiration. Inhibition of Complex V by oligomycin substantially inhibited the cybrids' respiratory activity, and the dependence of respiratory activity on translation of the transfected mRNA was supported by treating the cybrids with the mitochondrial protein synthesis inhibitor chloramphenicol, which completely arrested respiration.(10)
Treatment with the membrane-permeating JC-1 dye allows visualization and sorting of cells by the proportion of their mitochondria high (red) and low (green) mitochondrial membrane potentials (ΔΨm). 25% of untreated cybrid lines' mitochondrial populations were entirely composed of fully-depolarized organelles, and none of these cells had were enriched in high-ΔΨm mitochondria. Treatment with R8 alone did not affect the proportion of cells with fully-depolarized mitochondrial populations, though it caused some heterogeneity in the remaining cells' red staining; by contrast, treatment of cybrids with pcRNA1-R8 was able to fully restore the sorting pattern exhibited by mitochondrially wild-type HepG2 cells, and both cell types promptly exhibited high proportions of fully-depolarized mitochondria unpon treatment with an ETC uncoupling agent.(10)
Cells incompetent for OXOHOS depend entirely on glycolysis for ATP production and growth, and thus on glucose as an energy source, whereas OXPHOS-capable cells can generate ATP and maintain growth when deprived of glucose but supplied with galactose. In the presence of glucose, cybrid cells exhibited the same growth rate whether treated with pcRNA1-R8 or not. But provision of galactose alone to untreated cybrid cells did not permit growth and led to some cell death within 2 d, whereas cells transfected with the construct underwent 3 generations of replication, grew to confluence within 3 d, during which they maintained Complex IV activity. pcRNA1-R8 had restored OXPHOS to KSS cybrids to nearly wild-type levels, bypassing and effectively obviating the large deletions in the cells' own mtDNA.(10)
Conclusions
This in vitro experiment is exciting, opening up an alternative means of restoring oxidative phosphorylation to cells homoplasmic for OXPHOS-incompentent mitochondria with large deletions -- cells that accumulate in aging tissues, area associated with age-related diseases such as Parkinson's disease and sarcopenia, and that can strongly be argued to be contributors to the degenerative aging process. The method is rightly described by the authors as "inherently simple, efficient, and fast-acting, and appears to be of general applicability to a wide variety of cells and tissues (data not shown)." Indeed, it shows clear advantages relative to the low targeting to cells and/or mitochondria of previous attempts using pharmacological delivery systems, and appears to show a higher rate of restoration of OXPHOS and normal growth with galactose as the sole energy source than previous efforts using allotopic expression itself.
On the other hand, the effects of the RIC-based construct was transient, as would be predicted from the inherent nature of a therapy based on delivery of mRNA, which are routinely recycled within the cell: after the initial restoration of cybrid cell growth at day 3 of transfection, oxygen consumption rate declined to the basal levels over the ensuing 2 days, and cells began detaching from the vessel surface.(10) One would expect that repeated administration of "booster shots" would extend or restore OXPHOS in cells homoplasmic for mutant mitochondria in mitochondriopathy patients or persons who have undergone age-related mitochondrial deletions, but there is substantial room for skepticism that normal function could be continuously sustained by such means on acceptable booster schedules.
But this work is a major advance, and clearly promising. The therapeutic potential of the RIC-based should now be tested in animal models of inherited mitochondrial disease such as KSS, and if successful, the more ambitious work of using it to restore OXPHOS in the rare cells rendered homoplasmic for somatic mutations as a result of the degenerative aging of wild-type mice. The biomedical rejuvenation of aging human mitochondrial function would not lie far behind, with the promise of muscles maintained, Parkinson's prevented, and an end to the rising systemic metabolic toxicity of reductive cellular hotspots.(1)
References
1. de Grey AD. A mechanism proposed to explain the rise in oxidative stress during aging. J Anti-Aging Med 1998;1(1):53-66.
2.Khan SM, Bennett JP Jr. Development of mitochondrial gene replacement therapy. J Bioenerg Biomembr. 2004 Aug;36(4):387-93. Review. PubMed PMID: 15377877.
3. Mahata B, Mukherjee S, Mishra S, Bandyopadhyay A, Adhya S. Functional delivery of a cytosolic tRNA into mutant mitochondria of human cells. Science. 2006 Oct 20;314(5798):471-4. PubMed PMID: 17053148.
4. Mukherjee S, Mahata B, Mahato B, Adhya S. Targeted mRNA degradation by complex-mediated delivery of antisense RNAs to intracellular human mitochondria. Hum Mol Genet. 2008 May 1;17(9):1292-8. Epub 2008 Jan 18. PubMed PMID: 18203752.
5. Zullo SJ, Parks WT, Chloupkova M, Wei B, Weiner H, Fenton WA, Eisenstadt JM, Merril CR. Stable transformation of CHO Cells and human NARP cybrids confers oligomycin resistance (oli®) following transfer of a mitochondrial DNA-encoded oli® ATPase6 gene to the nuclear genome: a model system for mtDNA gene therapy. Rejuvenation Res. 2005 Spring;8(1):18-28. PubMed PMID: 15798371.
6. Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, Schon EA. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002 Apr;30(4):394-9. Epub 2002 Feb 25. PubMed PMID: 11925565.
7. Guy J, Qi X, Pallotti F, Schon EA, Manfredi G, Carelli V, Martinuzzi A, Hauswirth WW, Lewin AS. Rescue of a mitochondrial deficiency causing Leber Hereditary Optic Neuropathy. Ann Neurol. 2002 Nov;52(5):534-42. PubMed PMID: 12402249.
8. Bonnet C, Augustin S, Ellouze S, Bénit P, Bouaita A, Rustin P, Sahel JA, Corral-Debrinski M. The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim Biophys Acta. 2008 Oct;1783(10):1707-17. Epub 2008 May 6. PubMed PMID: 18513491.
9. Mukherjee S, Mahata B, Mahato B, Adhya S. Use of a parasite-derived protein complex to modulate the function of mitochondria in human cells. Rejuvenation Res. 2007 Sep;10(Suppl1):S19(Abs 2).
10. Mahato B, Jash S, Adhya S. RNA-mediated restoration of mitochondrial function in cells harboring a Kearns Sayre Syndrome mutation. Mitochondrion. 2011 Mar 23. [Epub ahead of print] PubMed PMID: 21406250.
View the full article
ImmInst
27 Apr 2011
SENS Foundation is hiring for our research center located in Mountain View, CA. We are seeking a team lead for our LysoSENS project, which contains both intra- and extramural components.
Qualified candidates will have an MS, or Ph.D. in the chemical/biological sciences and at least 5 years of work experience that must include prior project management experience. Duties will include the preparation of grant proposals, internal and external progress reports, individual and collaborative publication. The project lead will develop, interpret and implement standards, procedures, and protocols for the LysoSENS research program and may collaborate on determining strategic directions in the research program. Candidates must have a proven ability to lead other professionals.
Bench experience should include standard laboratory techniques, including but not limited to standard cell biology/biochem/molecular biology techniques. Good fundamental laboratory skills to include safety, microbial and mammalian cell culture. Duties may include cell culture and transfection, microscopy, protein production and analysis in addition to supervisory duties. Recombinant protein production in various systems, particularly yeast and bacteria, strongly desired. As a project lead, candidates must have the ability to design, develop and direct experiments that establish the viability of the SENS mission and chosen therapeutic goals.
Applicants should send their resume or CV to Tanya Jones (tanya dot jones at sens.org). Applications will be accepted through the end of May 2011.
View the full article
ImmInst
05 May 2011
SENS Foundation's Year End Report for 2010 is now available to download as a pdf. The Report includes: an overview of the year from our CEO, Mike Kope; a research summary from CSO Aubrey de Grey; commentaries on our Research Center operations and our outreach activities; and a breakdown of our 2010 finances.
View the full article
ImmInst
24 May 2011
Efficient, safe methods of gene therapy will be essential enabling technologies for the repair or obviation of several of the cellular and molecular lesions driving age-related disease and dysfunction, notably the accumulations of mutations in mitochondrial and nuclear DNA (including the medium-term obviation of the latter through WILT), as well as the introduction of novel lysosomal hydrolases to clear out age-associated intracellular aggregates.
As we've discussed in previous entries on the progress of gene therapy, zinc finger nucleases (ZFNs) are amongst the most promising methods under current investigation for human use. Even ZFNs, however, have some potential limitations. Perhaps the most important such limitation is that they rely on introducing double-strand breaks in the host genome, which are then repaired by exploiting the native Non-Homologous End Joining (NHEJ) DNA-repair machinery to insert an user-supplied DNA repair template for the novel gene. As such, the potential exists for even so high-precision a method as ZFNs to damage or disrupt non-target genes, introducing mutations or chromosomal aberrations. The potential is especially high in genes located at mutational "hotspots," whose sequence specificity or structural or functional features make them parti vulnerable to mutation in interaction with the repair and replication machinery of the cell.
These possible problems might ultimately manifest to a degree that proves unacceptable for direct use in somatic gene therapy; even in the case of cells modified ex vivo, where screening could potentially eliminate such abnormalities before therapeutic use, a high frequency of sporadic mutations would lower the net efficiency and power of the technique. Moreover, as with other nonviral approaches experiments to date, the use of ZFNs to modify the genomes of induced pluripotent stem cells (iPS) has thus far been characterized by low efficiency rates in what is already a low-efficiency technology; this is a significant limitation in itself, and might be further worsened if essential modifications of donor cells could only be achieved with methods associated with high rates of sporadic mutation.
In work just released in electronic form,(1) researchers working under Dr. Juan Carlos Izpisúa Belmonte at the Scripps Institute Center for Regenerative Medicine, have provided a strong proof-of-principle for the advantages of gene editing of iPS using helper-dependent adenoviral vector (HDAdVs), an approach that had already been shown to allow for efficient and precise gene editing in human embryonic stem cells, based on homologous recombination (HR).(4) This technology uses so-called "gutted" adenoviral vectors, generated by removing large sequences of viral DNA essential to the replication and packaging of the pathogen genome, and replacing their function with proteins from a helper virus or cell line in trans. This offers the simultaneous advantages of removing toxic or immunogenic viral proteins from the vector, eliminating the risk of mutations from double strand breaks, and opening up space for a large (~37 kB) payload of insert DNA.(2)
To test the efficacy of HDAdVs in introducing transgenes to iPS cells, the Scripps investigators chose as a strong test case a defective copy of the gene LMNA, which encodes lamin A, one of the proteins that make up the nuclear lamina, and which is thought to be involved in regulating gene expression, the stability of the nucleus, and chromatin structure. Inherited mutations in LMNA are responsible for Hutchinson-Gilford Progeria Syndrome (HGPS, commonly referred to as “progeria”); these mutations produce a truncated splicing defect of the protein, which accumulates and leads to nuclear defects including disorganization of nuclear lamina and loss of heterochromatin, resulting in a range of clinical signs and symptoms and early death in patients. While HGPS has been misleadingly characterized as “premature aging” on the basis a subset of the disease's phenotypes, there is likely no special need to target the gene as part of a panel of rejuvenation biotechnologies; however, its situation on a known mutational hotspot made it an excellent test target for HDAdV in iPS cells. Happily, Belmonte's group had recently established an iPS line from patient fibroblasts which harbors the mutant protein.(3)
High Efficiency
After transfection, the Scripps team evaluated the efficiency of integration using an inbuilt negative selection system activated by ganciclovir, screening out cells that had been subject to random integration. To their surprise, they found a high (78-100%) efficiency of integration using HR, even at very low multiplicities of infection, leading to successful integration of the transgene and correction of the mutation in 48% of HR cells.(3)
Successful Reprogramming Without Introduced Abnormalities
Importantly, the corrected HGPS-derived cells were by every test equivalent to wild-derived iPS. They exhibited a normal karyotype, expressed standard markers of pluripotency, had appropriate demethylation of the OCT4 promoter, and appeared to be pluripotent. Moreover, several tests of the HGPS-derived iPS vs. donor fibroblasts revealed no differences in single nucleotide polymorphisms (SNPs), no evidence of gene duplications or deletions; similarly, there were no detectable differences in gene expression profiles by DNA microarray analysis or of methylation patterns between HDAdV-corrected and -uncorrected HGPS-derived iPS.(3)
Phenotypic Rescue
However, LMNA is transcriptionally silent in iPS, so the correction of the mutant gene by the HDAdV system could only be demonstrated in differentiated cells derived from corrected HGPS-iPS. Accordingly, Belmonte's group differentiated the corrected HGPS-iPS into fibroblasts and smooth muscle cells (SMCs). These cells did not produce the truncated mutant lamin A protein, and did not exhibit the characteristic senescent phenotype of HGPS cells; this included a substantial reduction in staining with staining for senescence-associated beta-galactosidase (SAA-β-gal), which was reduced from 21.3% in SMCs derived from uncorrected HGPS-iPS to 6.8% in SMCs derived from corrected HGPS-iPS (comparable to the 11.4% in observed in wild-type BJ-iPS-derived cells), as well as a >60% reduction in the number of abnormal nuclei.(3)
Use in Adult Stem Cells
Buoyed by their success in correcting patient-derived iPS, the investigators tested the HDAdV vector in mesenchymal stem cells (MSCs), selected because the LMNA mutations responsible for HGPS most prominently affect mesoderm-derived tissues such as muscle and adipocytes, and because of MSCs' widespread use in early-stage regenerative medicine applications. Olfactory ectomesenchymal stem cells were chosen in particular to take advantage of their high proliferation rate, facilitating rapid clonal expansion. Wild-type cells were used due to the inavailability of HGPS-derived MSC. As with HGPS-iPS cells, the HDAdV system achieved high (54%) gene-editing efficiency in wild-type olfactory ectomesenchymal SC, and with no disruption of the native lamin A/C.(3)
A Contender for Use in Human Rejuvenation
HDAdV offers promise for the correction of genetic defects, and for the introduction of novel genes into cells for cell therapy and engineering new tissues impervious to the accumulation of a range of age-related cellular and molecular lesions. There is no reason to expect that a single system will be well-suited to all of the different gene therapy applications that will eventually form part of a comprehensive human rejuvenation strategy. The new system has specific strengths where high-efficiency introduction of large transgenes is required, or where loci of interest are especially vulnerable to disruption by other transgene vectors. Thus, either ironically or appropriately depending on one's point of view, this proof-of-principle in cells derived from a genetic defect commonly mistaken for an acceleration of the "normal" aging phenotype, may yet ultimately be harnessed to free aging persons from the dysfunction, disease and death that follow from the degenerative aging process.
References
1: Liu GH, Suzuki K, Qu J, Sancho-Martinez I, Yi F, Li M, Kumar S, Nivet E, Kim J, Soligalla RD, Dubova I, Goebl A, Plongthongkum N, Fung HL, Zhang K, Loring JF, Laurent LC, Izpisua Belmonte JC. Targeted Gene Correction of Laminopathy-Associated LMNA Mutations in Patient-Specific iPSCs. Cell Stem Cell. 2011 May 18. [Epub ahead of print] PubMed PMID: 21596650.
2: Mitani K, Graham FL, Caskey CT, Kochanek S. Rescue, propagation, and partial purification of a helper virus-dependent adenovirus vector. Proc Natl Acad Sci U S A. 1995 Apr 25;92(9):3854-8. PubMed PMID: 7731995; PubMed Central PMCID: PMC42060.
3: Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, Thompson J, Boue S, Fung HL, Sancho-Martinez I, Zhang K, Yates J 3rd, Izpisua Belmonte JC. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011 Apr 14;472(7342):221-5. Epub 2011 Feb 23. PubMed PMID: 21346760; PubMed Central PMCID: PMC3088088.
4: Suzuki K, Mitsui K, Aizawa E, Hasegawa K, Kawase E, Yamagishi T, Shimizu Y, Suemori H, Nakatsuji N, Mitani K. Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors. Proc Natl Acad Sci U S A. 2008 Sep 16;105(37):13781-6. Epub 2008 Sep 3. PubMed PMID: 18768795; PubMed Central PMCID: PMC2544531.
View the full article
ImmInst
01 Jun 2011
Max Peto joined the SENS Foundation Research Center team as a Researcher in June 2010. He has since been working on the expression of A2E-degrading enzymes for the Center’s LysoSENS project. A2E is suspected to be the primary cause of age-related macular degeneration, a disease that occurs when the molecule accumulates in the lysosomes of retinal pigment epithelial (RPE) cells in the eye. Most recently, Max has been optimizing growth conditions and media composition for yeast cultures, expressing and purifying A2E-degrading enzymes, performing western blots and activity assays, and purifying A2E that he had previously synthesized.
For Max, working at the SENSF-RC has been the culmination of years of dedicated study and preparation. Before he first heard about SENS in early 2005, he wasn’t a scientist at all; in fact, he was a 23-year-old cost accountant. When he wasn’t studying for his MBA, he was counting other people’s money. He knew that he wanted more out of life, though: specifically, he wanted to change the world in a way that would benefit society. As soon as he found and read Aubrey de Grey’s Ending Aging, he settled on human health as the area he would strive to impact-- and on SENS as the way to make that impact. Over the next few years he committed himself to working in finance, at one point teaching at a local community college, always with the intention of saving his money so that he could return to school to learn about science and laboratory work. During this period he studied whenever he had the time, reading articles relevant to health and aging in scientific journals.
In 2008, Max went back to school full-time at the University of Toledo to study chemistry, math, and biology. He was interested in taking an active role in SENS research as quickly as he could, so he contacted the predecessor of the SENS Foundation Academic Initiative, MFURI. As a member of the Initiative, he performed a literature review on the harm caused by iron and aluminum accumulation in the body, citing well over a hundred journal articles. Max’s paper was accepted by the journal Rejuvenation Research and published in April 2010, just as he was completing his coursework at Toledo. As his next step, Max opted to join the RC staff rather than pursue a PhD opportunity so that he could continue to make as direct and immediate of a contribution to SENS as possible.
Max has now been working at the SENSF-RC for one year, and will be staying on to continue his work on the A2E degradation project. In the long term, he hopes to see the LysoSENS project through all of its pre-clinical stages. It is his wish that this work will lead to therapies that can effectively reverse, or at the least greatly slow, the pathology of age-related macular degeneration.
View the full article
ImmInst
22 Jun 2011
As we've noted previously,
Neurofibrillary tangles (NFT -- cytoplasmic inclusions composed of phosphorylated and abnormally-cleaved species of tau protein) accumulate in the aging brain, and at higher levels in Alzheimer's disease and in vulnerable regions in a range of other neurodegenerative diseases; they are closely associated with neuronal death and with onset of clinical dementing disease. The clearance of neurofibrillary tangles and other intracellular aggregates is a key rejuvenation biotechnology to restore aging brain function.
The priority of a distinct therapy for the removal of tau pathology has become especially clear in light of followup studies in persons receiving the original, active beta-amyloid [Aβ] vaccine AN1792. On the one hand, vaccine responders' brains exhibited a nearly complete absence of Aß pathology at autopsy, along with reduced neuronal loss, and in long-term (4.6 y) followup, a decline on the Disability Assessment for Dementia and Dependence Scale, stabilized hippocampal volume while adjuvant-only controls suffered ongoing declines, and extensive clearance of tau-containing neurites.[references] Yet narrowly cognitive benefits were limited, and more mature tau pathology (NFT and neuropil threads) appeared to be unaffected ([references], and see previous postings).This last finding, combined with the stronger association of NFT burden with clinical disease, recommend NFT clearance as a high-priority (and, ideally, complementary) immunotherapeutic approaches.
In three previous posts, we've discussed progress by on this front by Dr. Hanna Rosenmann's group at Hadassah University Hospital, Israel, and Dr. Einar M Sigurdsson and colleagues at New York University, who have tested immunotherapies targeting tau aggregates in preclinical models of neurodegeneration caused by pathological tau species. Heretofore, they have worked with transgenic models express either wild-type human tau or highly aggregation-prone mutant tau species in addition to the native murine tau gene; in such models, they have demonstrated that, somewhat surprisingly, active vaccination with tau species generates tau-targeting antibodies that not only cross the blood-brain barrier, but clear out intraneuronal pathological tau species, via a presumed lysosomal mechanism; in so doing, they have achieved significant reductions in tau pathology and the associated functional deficits. These findings not only support the importance of tau as a therapeutic target, but offer grounds for optimism for the development of tau-targeting rejuvenation biotechnology for human beings.
Amongst the limitations of these findings, however, has been the nature of the model organisms themselves. Some of these models express mutant tau species such as P301S and K257T, which (respectively) cause the human dementing disorder frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) and a murine tauopathy similar to frontotemporal dementia (FTD). However, these mutated tau species are not present either in Alzheimer's disease, or in "normal" brain aging (mistakenly so-called). While other models have instead shown similar pathology through transgenic expression of wild-type human tau (htau) superimposed on the native murine tau (and similar benefits from removing it), all such models have to varying degrees shared the significant disadvantage of causing such severe, widespread, and early-onset tau pathology that motion disorders have significantly confounded the evaluation of cognitive function, further emphasizing the limited translatability of the models and introducing uncertainty over the degree to which true cognitive deficits are being induced or alleviated.
Now, Sigurdsson's group have made a report further advancing the case for the therapeutic importance and tractability of pathological tau, by demonstrating similar robust benefits to immunotherapy murine model of tau-based cognitive deficits that are not confounded by brainstem involvement or movement disorders.
The main model in the new study(1) was developed by crossing mice expressing transgenic wild-type htau with a model carrying the human presenilin 1 (PS1) mutation M146L, on a murine tau knockout background. "These mice have an earlier onset, at or before 2 months of age, and more rapid progression of tau pathology than the htau mice, while the distribution is similar, with extensive involvement of hippocampal and cortical regions"(1) rather than being concentrated in the brainstem and spinal cord as in previous models. PS1 mutations accelerate tau pathology, the authors suspect, by impairing lysosomal function.(2) These animals develop robust tau pathology consistent with that seen in human Alzheimer's disease, and exhibit cognitive deficits on several established tests (radial arm maz, object recognition tes, and closed field symmetrical maze) but no impairments of motor function (as evaluated by rotarod, radial arm maze, or traverse beam) -- features that make this model especially promising for the screening of potential tau-targeting therapies.
Sigurdsson's group developed a vaccine based on Tau379–408[P-Ser396, 404], "selected based on its overall immunogenicity and its AD phospho-epitope" and administered "intraperitoneally in 100 μl of alum adjuvant (Adju-Phos, Brenntag Biosector)".(1) The htau/PS1/mtauKO mice were first immunized at 3-4 mo of age, significantly after the 2 mo disease onset time, followed by 3 biweekly injections and then further booster immunizations monthly thereafter. These animals were compared not only to adjuvant-only htau/PS1/mtauKO mice, but also to adjuvant-only htau/PS1/mtau and htau/mtauKO mice "as additional controls because our preliminary analysis indicated that these models had less pathology than the htau/PS1 [+ mtau knockout] model. ... At 7–8 months, the mice went through extensive behavioral testing and were subsequently killed for tissue analyses at 8–9 months of age."(1)
Vaccinated mice developed strong IgG antibody titers targeting both the phosphorylated tau immunogen and unphosphorylated htau, "As expected, because of the overall high immunogenicity of the immunogen ... although a better response is initially generated against the phosphorylated immunogen. Recombinant tau is recognized as well in both controls and immunized mice but to a much lesser degree than the immunogen epitope ... Some autoantibodies are detected in controls and are likely also present in the immunized mice. ... IgM response was less pronounced, as expected, but was of a similar pattern as the IgG response. Plasma from the immunized mice recognized tau pathology in AD and mouse tissue (data not shown), as we observed previously with this immunogen in [P301L tangle model mice]"(1)
Vaccination with human phosphorylated tau led to the clearance of tau pathology from the brains of immunized mice, as revealed using both immunohistochemical staining with antibodies to the phosphorylated tau species PHF1 and AT8, and Western blot analysis of total and phosphorylated tau species (normalized with actin and total tau levels), evaluated using (respectively) polyclonal B19 antibody, and with monoclonal antibodies to PHF1 and CP13.

Figure 1: Phospho-tau immunotherapy lowers brain pathological tau burden. Reproduced from (1); see larger image at publisher website.
staining with PHF1 and AT8 antibodies revealed pronounced tau pathology, primarily in the htau/PS1 controls and to a lesser and comparable degree in the other three groups ... Further analysis indicated a very strong trend for the immunotherapy to reduce the ratio of PHF1/actin by 35 and 42%, respectively, in the soluble and insoluble fractions. ... The therapy reduced PHF1-reactive tau aggregates by 57% in the pyriform cortex (p < 0.01), ... [a] brain region chosen for analysis in the htau model because of its prominent pathology ... compared with identical controls. ... However, the immunotherapy appeared to reduce tau pathology throughout the brain. ... The regional pattern of tau pathology was similar as described previously for the htau model, with prominent cortical and hippocampal involvement, but was more severe in the htau/PS1 model at the age analyzed. A time-course study of the progression of brain pathology in the htau/PS1 model is underway.(1)
In turn, these reductions in abnormal tau species were clearly linked to substantial improvements in cognitive deficits, on all three tests:

Figure 2: Immunological and cognitive testing results in vaccinated htau/PS1/mtauKO mice, and multiple adjuvant-treated controls. Reproduced from (1); see larger figure at publisher website.
Importantly, the cognitive improvements correlated well with reduction in PHF1-stained tau aggregates assessed by immunohistochemistry. Significant correlation was observed in all three memory tests ... Less consistent correlations were observed between the Western blot fractions and cognitive outcomes that varied depending on the fraction (soluble, insoluble) of tau antibody (PHF1, CP13), the protein used for normalizing the data (total tau, actin), and the cognitive test (data not shown). These findings indicate that tau pathology on histological sections rather than Western blots may predict cognitive outcome. Overall, these results strongly demonstrate the feasibility of tau immunotherapy for AD and related tauopathies.(1)
This is an impressive advance. The authors have used vaccination with a human phosphorylated tau immunogen to effect the immunologic clearance of pathological tau aggregates associated with Alzheimer's disease, in a mouse model expressing wild-type human tau. They have intervened late, months after the initial development of cognitive deficits. In using transgenic wild-type presenelin, they may have added early lysosomal deficits similar to human AD (2) which impair the normal autophagic clearance of wild-type and pathological tau species. For the first time, the model exhibits cognitive deficits that are both secondary to the accumulation of pathological tau species, and (to use a slight oxymoron) "clean," being unconfounded with the motion disorders that constituted a significant caveat to the relevance of previous models. And the vaccine has not only elicited a robust immunological response, and cleared pathological tau species from brain regions of relevance to human disease, but have linked such clearance to improved cognitive function on several extensively-used tests.
The new work is strong support for the therapeutic importance and tractability of the removal of pathological tau species from the brain -- in Alzheimer's disease, in other tauopathies, and in the "normal" degeneration of the aging brain. And it is yet another in a mounting series of reports offering support for the therapeutic heuristic of removing the damage of aging, to effect the rejuvenation of the body -- and the mind that coincides with its structural and functional integrity.
References
1: Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010 Dec 8;30(49):16559-66. PubMed PMID: 21147995.
2: Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010 Jun 25;141(7):1146-58. Epub 2010 Jun 10. PubMed PMID: 20541250.
View the full article
ImmInst
07 Jul 2011
I recently travelled to Zaragosa, Spain, to attend the eighth European Meeting on Mitochondrial Pathology (Euromit 8). The conference was extremely relevant to the MitoSENS project, and I was very lucky to have the opportunity to attend.
For me, the most striking aspect of this scientific conference was the cross-sectional interaction between clinicians and basic scientists. There were medical doctors describing symptoms of mitochondrial diseases, reporting on clinical trials, and proposing new ideas for treatments. I have never met so many MDs who were so interested in the basic science of a cellular component. It was interesting to see how much the basic scientists relied on data from patients to learn about the function of mitochondrial genes, by studying the patient symptoms and utilizing mutant tissues and cell lines derived from them. We at the SENSF-RC are going to be relying on some such cell lines in the near future.
One inspiring story was of a treatment for a disease called MNGIE (mitochondrial neurogastrointestinal encephalopathy) which is both debilitating and fatal. The gene mutated in this disease (thymidine phosphorylase) is a secreted enzyme which prevents toxic levels of thymidine from building up in the body. “By golly” you exclaim, “secreted enzyme deficiencies can be treated using enzyme replacement therapy!” Well, the researchers (data presented by Dr. Michio Hirano) thought the same thing, so they approached drug companies to ask them to make it. The drug companies were interested — until they found out how few patients there are to treat and how little money they would make from such a treatment. The clinical researchers pressed on, however, with an alternative approach. They hypothesized that normal cells from a healthy donor would secrete some enzyme that could help the patients. They were right; after successful bone marrow transplants, many patients made dramatic recoveries. The researchers are now working on refining the procedure so that they can help more people, and treat everyone who suffers from this debilitating disease.
Euromit was a great opportunity to interact with researchers relevant to the MitoSENS project - in particular, with Marisol Corral-Debrinski and her fantastic research group. It is her technique of co-translational import of allotopically expressed proteins upon which our current efforts are built. I shared our progress over the past 6 months with her, we talked shop, and she had some great suggestions. Most importantly, she seems to think we are on the right track, and her own research is progressing most impressively. It will be very exciting to witness the progress of her clinical trials treating blindness caused by LHON over the coming months and years.
I returned from Spain energized and motivated to conquer MitoSENS once and for all!
View the full article
ImmInst
06 Aug 2011
As we have previously reviewed, a comprehensive suite of rejuvenation biotechnologies must include the removal of extracellular aggregates from aging cells and tissues. The most clinically-advanced such biotechnology is immunotherapy against aggregated beta-amyloid protein (Aβ), a characteristic neuropathological lesion that accumulates in the brain with age and contributes to cognitive decline during "normal" brain aging and dementia in Alzheimer's disease (AD).(0)
While Aβ was originally identified neuropathologically in the form of the characteristic plaques that accumulate in the AD brain, the view that soluble, intracellular oligomeric Aβ species are of equal or greater pathological significance has emerged and become dominant over the course of the last decade.(1-4) These Aβ species may be generated intraneuronally, or may be internalized from extracellular Aβ; irrespective of origin, evidence exists that the degradation of Aβ of either type may rely upon lysosomal hydrolysis, in both the neuron(2) and microglia,(6) and more surprisingly, that Aβ-targeting antibodies act via cellular uptake, followed by trafficking bound Aβ into the lysosome for disposal.
Dr. Ben Bahr and his colleagues with the Neurosciences Program and Division of Pharmaceutical Sciences at the University of Connecticut have for some time now been investigating the effects of elevating lysosomal activity using the lysosomal modulator Z-Phe-Ala-diazomethylketone (PADK). PADK had previously been shown to reverse the AD-like abnormalities of the induced in hippocampal slices treated with the lysosomal inhibitor chloroquine(7):
In the hippocampal slice model, tau deposits and amyloidogenic fragments induced by the lysosomal inhibitor chloroquine were accompanied by disrupted microtubule integrity and by corresponding declines in postsynaptic glutamate receptors and the presynaptic marker synaptophysin. In the same slices, cathepsins B, D, and L, beta-glucuronidase, and elastase were upregulated by 70% to 135%. ... [C]hloroquine was applied for 6 days after which its removal resulted in continued degeneration. In contrast, enhancing lysosomal activation by replacing chloroquine after 6 days with PADK led to clearance of accumulated protein species , [restoration of synaptic composition(8)], and restored microtubule integrity.(7)
Similar lysosomal-endosomal, axonal, and synaptic pathology was later reported in the same model using Aβ1–42,(8) and in vivo, intraperitoneal injections of PADK in rats were also shown to dose-dependently elevate forebrain levels of the lysosomal proteases cathepsin D by 50–100%, and cathepsin B by 40–80%.(8)
In a new study, Dr. Bahr's group has extended this work into a transgenic mouse model of AD, testing PADK's ability to retard, and to reverse, AD neuropathology and cognitive dysfunction in two models of transgenic AD mice: 10-11-month old APP(SwInd) mice, which express a relatively low level of the parent strain transgene copy number and accumulate relatively low levels of Aβ neuropathology, and 20–22 month old APPswe/PS1ΔE9 mice, with greater and longer-established burdens of Aβ. (6)
As in the hippocampal slice model, cathepsins B and D are upregulated in AD and aging brain, evidently to counteract rising levels of interneuronal Aβ; however, older APP(SwInd) mice fail to exhibit such an upregulation, suggesting that homeostatic responsiveness can be overcome .at high levels of proteotoxicity and/or that such a failure contributes to further degeneration in the older animals. Systemic PADK injection of PADK in both models caused 3- to 8-fold increases in cathepsin B levels and similar elevations in the enzyme's activity in lysosomal cell fractions, but did not affect other Aβ-degrading enzymes (neprilysin and insulin-degrading enzyme). Vessicle-trafficking Rab protein levels were altered in vitro, without an effect on the lysosomal membrane protein LAMP-1, an essential receptor for chaperone-mediated autophagy.(6)
Accordingly PADK-induced lysosomal modulations cleared a significant amount of the intra- and extraneuronal burden of Aβ from treated mice, reducing intraneuronal Aβ regions of the hippocampus and piriform cortex by 63–73% in younger mice and by ~50% in older ones; in the latter animals, extracellular Aβ was concommitantly reduced by an impressive 76–85% (see Figure 1, below). Contrariwise, PADK had no effect on recombinant human BACE1 activity, and cleavage products of α- and β-secretase were unchanged, weighing against modulation of Aβ production. Even as PADK lowered levels of the highly neurotoxic Aβ(1-42) species, it elevated levels of the less pathogenic Aβ(1-38), suggesting that PADK-induced elevation of cathepsin B truncated Aβ(1-42) levels into a safer cleavage product.(6) These results are consistent with an earlier study,(9) not cited by the authors, which reported that virally-mediated overexpression of cathepsin B in aged model AD mice caused significant recession of existing plaque, associated with the production of less-amyloidogenic truncated Aβ peptides.
Figure 1: PADK decreases intra- and extraneuronal Aβtargeting antibody staining in aged
APPswe/PS1ΔE9 mice. Reproduced from (6)
Model AD animals exhibited evidence of synaptic degeneration, in the form of 23–31% reductions in the synaptic markers synapsin II, synaptophysin, and GluA1 relative to wild type; PADK completely restored normal levels of GluA1, in both young and old AD mouse cohorts, and had similar effects on synapsin II and synaptophysin. "The integrity of hippocampal dendritic fields was also found preserved in immunostained tissue sections, and the level of GluA1 immunoreactivity within each transgenic mouse correlated with the respective extent of cathepsin B enhancement in the brain."(6)
As expected, model AD mice also exhibited substantial impairment of performance on cognitive-behavioral tests including the suspended rod, exploratory habituation, and spontaneous alternation behavior in a T-maze tests. PADK-induced Aβ clearance resulted in the complete restoration of normal function in both young and old animals (Figure 2).
Figure 2: Rejuvenation of cognitive function in AD model mice. Reproduced from (6).
The results are surprisingly robust. Through a simple additive stimulation of the existing compensatory upregulation of the native lysosomal hydrolytic machinery, intra- and even extraneuronal Aβ burden was substantially cleared, even in old animals with well-established disease. Moreover, the clearance was accompanied by the repair of disease-associated synaptic pathology, and a thorough rejuvenation of cognitive function. As a tribute to the power of the cellular waste-disposal machinery, these results are impressive.
Equally, they are not a solution to human brain aging. These animals, like most transgenic models of AD, exhibit no tau pathology nor significant neuronal loss -- problems that will also have to be addressed in order to achieve the full prevention of AD and rejuvenation of aging brains. Constant stimulation of autophagy would be expected to lead to impaired protein synthesis, possibly contributing to sarcopenia, thrombocytopenia, or impaired wound healing or recovery from exercise, injury, or illness. Moreover, overexpression cathepsin B "has been associated with esophageal adenocarcinoma and other tumors." But they do point to the potential of fortification of the lysosomal hydrolytic complement as one key element of a comprehensive suite of rejuvenation biotechnologies for the brain, as a complement to the inadequate native complement's ability to degrade Aβ in neurons and microglia, and as a complementary strategy to maximize the effectiveness of Aβ-targeting immunotherapy.
References
0: Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
1: Ono K, Yamada M. Low-n oligomers as therapeutic targets of Alzheimer's disease. J Neurochem. 2011 Apr;117(1):19-28. doi: 10.1111/j.1471-4159.2011.07187.x. Epub 2011 Feb 9. Review. PubMed PMID: 21244429.
2: Tampellini D, Gouras GK. Synapses, synaptic activity and intraneuronal abeta in Alzheimer's disease. Front Aging Neurosci. 2010 May 21;2. pii: 13. PubMed PMID: 20725518; PubMed Central PMCID: PMC2912028.
3: Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008 Apr 16;28(16):4231-7. PubMed PMID: 18417702; PubMed Central PMCID: PMC2685151.
4: Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007 Jun;101(5):1172-84. Epub 2007 Feb 5. Review. PubMed PMID: 17286590.
5: Butler D, Hwang J, Estick C, Nishiyama A, Kumar SS, Baveghems C, Young-Oxendine HB, Wisniewski ML, Charalambides A, Bahr BA. Protective effects of positive lysosomal modulation in Alzheimer's disease transgenic mouse models. PLoS One. 2011;6(6):e20501. Epub 2011 Jun 10. PubMed PMID: 21695208; PubMed Central PMCID: PMC3112200.
6: Yang CN, Shiao YJ, Shie FS, Guo BS, Chen PH, Cho CY, Chen YJ, Huang FL, Tsay HJ. Mechanism mediating oligomeric Aβ clearance by naïve primary microglia. Neurobiol Dis. 2011 Jun;42(3):221-30. Epub 2011 Jan 8. PubMed PMID: 21220023.
7: Bendiske J, Bahr BA. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis--an approach for slowing Alzheimer disease? J Neuropathol Exp Neurol. 2003 May;62(5):451-63. PubMed PMID: 12769185.
8: Butler D, Brown QB, Chin DJ, Batey L, Karim S, Mutneja MS, Karanian DA, Bahr BA. Cellular responses to protein accumulation involve autophagy and lysosomal enzyme activation. Rejuvenation Res. 2005 Winter;8(4):227-37. PubMed PMID: 16313222.
9: Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, Gan L. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron. 2006 Sep 21;51(6):703-14. PubMed PMID: 16982417.
View the full article
ImmInst
10 Aug 2011
SENS Foundation, a Californian non-profit, seeks an IT manager to work as part of its expanding team. The position is 2.5 days per week.
About SENS Foundation: SENS Foundation is a non-profit, life sciences organization with a mission to develop, promote and ensure widespread access to rejuvenation biotechnologies which comprehensively address the disabilities and diseases of aging. Our Research Center in Mountain View, California is the hub for a growing team of researchers, and outreach and executive staff. Several members of staff are located outside California, in the USA or Europe. In addition, we award grants to affiliated universities and research organizations.
About the position: The IT Manager will, in the first instance, report to our Director of Research Operations, Tanya Jones. However, the successful candidate will be required to work with all members of staff to consolidate and expand our IT infrastructure, both internal and public-facing. Initial projects will include: migration of email systems from legacy servers; implementation of a collaboration framework for files and documents; a review of our web systems, their integration with finance and customer relations back-ends, and associated social networking sites.
About the candidate: The successful candidate will have proven abilities in the technical aspects of the position, detailed below. He or she will also have experience with project planning, including gathering user requirements, performing software and hardware comparisons, creating functional specifications, implementation plans and costings, managing a budget, etc. The position requires an ability to understand the key business requirements at all levels of the organization, whilst demonstrating independent management skills at the IT project level.
In addition to the above, the position has the following requirements:
- 4+ years of experience in local and remote support of end users and infrastructure in mixed-platform environments, including Windows, Linux, OS X and associated filesystems and protocols
- Experience in planning, deploying, and testing backup/disaster recovery systems
- Experience of most, if not all, of the following environments and websites (or close equivalents): Google Apps, QuickBooks, SalesForce, Facebook, Twitter, YouTube, Paypal
- Experience in developing and deploying Web applications, ideally using Drupal
- A good grasp of networking fundamentals (cabling, switching, routing, TCP/IP) and common Internet protocols (HTTP, SMTP, etc.)
- Very strong organizational skills with the ability to manage multiple concurrent projects and support requests
- Good verbal and written communication skills
- A self-starter attitude coupled with a strong desire to work within a tightly-knit team
The following, although not essential, are also desirable:
- Familiarity with ticketing systems
- Fluency in a scripting language (Perl, Python, PHP, etc.)
- Experience in a scientific/laboratory environment
View the full article
ImmInst
16 Aug 2011
The SENS Foundation Academic Initiative’s new structure is actively in the process of being implemented, and involves a number of significant changes. Among these are the separation of the Initiative into branches, an updated membership system that allows students to become involved more easily and in more ways, the creation of volunteer committees, and the addition of outreach projects to the Initiative’s activities. A brief summary of these changes follows.
Branches
There will be three branches: Research, Outreach, and Education. The Research branch will be focused on the actual accomplishment of scientific research. This research will always be done with an eye to publication, but its most important function will be to provide our students with learning experiences, to help them develop into career scientists. The Outreach branch will be focused on spreading the word about the Academic Initiative and about the SENS Foundation, while the Education branch will be focused on educating students about science and SENS.
Membership
People involved with the Academic Initiative will be divided into three groups: staff, volunteers, and members. Staff will be employees of the SENS Foundation, while volunteers will be people who serve on committees that support and assist our students. Members will be students, mentors, and even professors: those who might be actively engaged in completing research, doing outreach, or learning about SENS. Students will no longer need to have a research project in order to join the Initiative, so we will be able to integrate more members into the program.
Committees
Volunteer-based committees that are already active include the Literature Review Guidance Committee, which guides and assists students who are working on literature reviews, and the Web Recon Committee, which searches for valuable online resources that can help our students. Other committees that will be forming include the Editorial Committee, which will review student manuscripts and provide feedback, and the Campus Outreach Committee, which will help to create outreach projects and foster the development of student groups.
Outreach Projects
While the Academic Initiative has long helped students to complete research projects, it has not done much in the past to encourage students to be advocates of the Initiative and the SENS Foundation. This will change with the implementation of outreach projects. These will generally be simple, off-the-shelf projects that students can finish in an afternoon, such as printing fliers from a pre-made template and distributing them at their university. These projects will not be considered to be entirely complete, though, until their impact has been analyzed, such that we might learn what methods are most effective.
New Policies
The Academic Initiative is implementing and will implement a number of new policies. These include a “Project Registration” policy that will allow students to formally “register” their projects with the Initiative, that we might bring more rejuvenation-related projects into the Initiative, even if these projects themselves do not come from ideas generated by SENSFAI mentors. An “Open Phone” policy will also go into effect, such that members of the Initiative will always be able to contact the Academic Coordinator. Finally, all members will have a voice chat with the Academic Coordinator upon entry to the Initiative. This last policy has already been implemented retroactively.
More updates and changes are in the works. If you would like to stay up-to-date, you can join the Academic Initiative’s email list here.
We can always use more volunteers, students, mentors, and researchers. If you would like to become involved with the Initiative, you should contact the Interim Academic Coordinator, Daniel Kimbel, at daniel dot kimbel at sens dot org.
View the full article
ImmInst
18 Aug 2011
As a part-time Laboratory Hardware Specialist here at the SENS Foundation's California-based research center, I can often be found peering into the innards of one machine or another, attempting to solve age-old mysteries along the lines of "Why won't this thing turn on?" or "It was working five minutes ago!" Which brings me to the inspiration for this post: that humble device known as the centrifuge. Centrifuges are of major importance in biotech research.
One might even say they make the world of cells, genes, and DNA go around.
Ahem.
Moving along, currently we've got the following six centrifuges here at the RC:
(1) Revolutionary Science (microcentrifuge)

(2) Labnet Hermle Z 233 M-2

(3) Hill IMV-15 MicroCentrifuge / IBI A Kodak Company

(4) Fisher Scientific AccuSpin Micro 17R

(5) Sorvall RT-7 Plus

(6) Sorvall RC-5B

Currently all of these units (except the RC-5B, which was acquired most recently) are in working order, and most are subject to frequent and vigorous use. I must admit this has been somewhat of a surprise to see -- prior to getting such a close look at biotech research in action, I probably would have figured a centrifuge was a centrifuge was a centrifuge. And oh, how wrong I would have been!
The more time I spend in this environment, the more it becomes clear that every model in fact provides something very specific. Centrifuges can differ in everything from the size and number of tubes they accommodate, to their temperature control and refrigeration abilities, to the range of speeds they can run at.
Moreover, many centrifuges (particularly the larger ones) can be equipped with a wide range of rotors in order to suit various purposes and requirements. The two main types of rotors you'll likely see in most labs are fixed angle type and hanging bucket type.
Fixed angle rotors resemble a cone with the pointy end lopped off and holes for test tubes arranged around the center (where the rotor connects to the spinning shaft inside the centrifuge). It is easy to understand the reason for the existence of these two rotor types when you picture the contents of a tube in the centrifuge while it is spinning. If the tube is held at a fixed angle, the contents will be pulled into an oblique shape along the length of one side.
If the tube is situated in a hanging bucket, however, the bucket will swing out toward an angle parallel with the horizontal plane. In this latter case, the denser contents will be forced through the "full depth" of the fluid toward the bottom of tube.
In other words, each of the two primary rotor types has its own set of advantages mainly due to its effect on tube contents. E.g., you can often expect separation to occur more rapidly in the fixed-angle case, as the denser parts of the contents only have to travel a short distance through the fluid before reaching the side of the tube (and sliding to the bottom, in cases where the particulate being separated is significantly more dense than the medium in which it is suspended, e.g., when "pelleting" bacteria or yeast).
This is fine and quite desirable in many instances, which -- in addition to their being fairly low maintenance due to a lack of moving parts -- is why fixed-angle rotors are so common.
On the other hand, sometimes the contents of a tube will include multiple parts to be separated, and it will not be possible to distinguish between those parts on the basis of density unless you can establish a strong force vector along the entire length of the tube. In the aforementioned bacteria/yeast suspension case you've got quite a different situation than that of, say, a tube full of cell matter (following homogenization of the cells).
In the cell-matter scenario you're not just separating solid from liquid, but multiple solids from other solids. Which brings me to the reason we've not been able to use the big RC-5B centrifuge just yet: basically, the rotor we need (an SH-80 hanging-bucket unit) is proving rather elusive.
We got this centrifuge in the first place because it can run at speeds fast enough to perform the "higher resolution" separations of multiple minute particles required by certain experiments. Cell fractionation, for instance, could be performed far more quickly and feasibly here with a functioning ultracentrifuge -- the idea here is that you want to end up with an "organelle parfait", wherein each layer represents a different type of organelle. And while we've located a few secondhand SH-80 rotors for sale, unfortunately thus far it seems that everyone who has ever owned one of these things has mysteriously lost all the buckets.
All that aside, even if we did find a used SH-80 with all its parts intact, we would need to be very careful. When it comes to rotors, you really need to ascertain that they are in good condition and not suffering from mechanical wear or other damage. Otherwise you risk ending up with, for instance, a rotor that splits in half during use due to metal fatigue.
It is also critical to use the correct rotor for a particular model, rather than attempting to "kludge in" one that seems "close enough". As an inveterate kludger it has been something of a task coming to terms with this, but given that the consequences of using the wrong rotor can include explosion of the centrifuge itself, damage to other nearby equipment, and shockwaves of literally window-shattering magnitude, there is really no room to view large centrifuges as anything other than really serious business.
In other words, it is vitally important in a job like this to know when to just cut your losses and find an alternative. We aren't quite ready to give up on the RC-5B here just yet, but if it turns out that our only option is a brand-new rotor (which would cost far more than the centrifuge itself did), we're going to have to look elsewhere.
On a more cheerful note, most of the other centrifuges here are reasonably user-serviceable, which is good considering they've all needed some sort of special attention over the past year. I've certainly gotten some nice opportunities to utilize my electrical engineering background, given that motors, electromechanical latches, sensors, etc., tend to figure prominently in centrifuge operation. The most commonly observed basic symptoms we see here ("it won't spin", "it won't turn on", etc.) can stem from anything from a burned-out power supply to a broken wire to a bad sensor to probably lots of other erroneous conditions I've yet to meet first-hand.
Power supply issues seem to be very common in lab equipment in general, so that's become one of the first places I look when something seems amiss. Consumer and industrial electronics are generally built to withstand certain transient levels, but if their protection components fail due to factors like age, accident, or manufacturing defect, even "normal" facility electrical supply behavior can impact power supply circuitry in various not-very-nice ways.
For instance, our little Revolutionary Science microcentrifuge was brought to me earlier this year in a sad state of having "let the magic smoke out". I opened the unit, and beyond the rather alarming burn marks on the printed circuit board within, I could see that the power supply circuitry was very, very simple. Which was fortunate because it meant I was able to de-solder and replace the rectifier which looked to have been destroyed by a transient voltage spike. Hooray!
On the other end of the spectrum, occasionally the solution to an initially puzzling problem turns out to lie in adjusting the machine's environment rather than the machine itself. E.g., a few months ago the lock mechanism on our second-largest refrigerated unit (the Sorvall RT-7 Plus) started refusing to engage. Most centrifuges won't spin unless their lid is properly latched down (for obvious safety reasons), meaning that until we could get the RT-7 to lock, it was useless.
Needless to say, there was no time to waste -- while the RT-7 Plus isn't massive, it does accommodate the larger (45 mL) test tubes, which are useful for speeding things up whenever a larger volume of experimental media requires processing. When the malfunctioning lock was brought to my attention the first thing I did (well, after making sure everything was unplugged!) was remove part of the RT-7's chassis. This revealed the nature of the unit's electromechanical locking mechanism: that is, certain metal parts needed to be able to contact other metal parts in order for the lock to actually engage.
Initially I thought the mechanical bits might need re-oiling or something along those lines, but then I saw the problem and nearly laughed aloud: the lock mechanism was actually being physically blocked by a piece of cardboard someone had folded up and shoved under the front "foot" of the centrifuge in order to level it. Sure enough, removing the cardboard restored lock functionality, which in turn rendered the RT-7 usable once more. Hooray again!
While this is by far not the end of our adventures with centrifuges here at the SENS RC, it is a good point to end this post. Thanks for reading, and hope you'll stop by again for more assorted tales of equipment, experiment, and other fascinating aspects of lab life!
View the full article
ImmInst
16 Sep 2011
On 19 September, 2011, Sarah Marr will be stepping down as our Executive Vice President at SENS Foundation. She has been a committed co-founder, and she will of course continue to be a trusted advisor and closely involved with the organization. But we couldn't have her term of full-time service with us pass without noting the significant contribution she has made to the professionalism of the organization and to the quality of our overall message. She helped make us, in a very real way.
I've known Sarah as an energetic colleague with a mind for both the overall strategy and all the pesky details to achieve it, and I've known her as a great friend. I'll keep the latter, but sure will miss the former.
(Sarah has written a short post on her decision on her personal blog.)
View the full article
ImmInst
28 Sep 2011
Sarah Fazal joined our research center team as an intern for the summer. Over the past few months, she worked with our MitoSENS team, primarily verifying the integration of DNA transfected into cells and detecting RNA expression levels. Her efforts contributed greatly to the progress our MitoSENS team has made over recent months, and she presented those results in a poster at our recent SENS5 conference in Cambridge. We'd like to thank Sarah for all her hard work over the past few months and wish her well in her graduate studies; and now, without further ado, here is her internship report:
 I clearly remember my first day at the SENS Foundation research center in Mountain View, California. It was a gorgeous morning with clear skies and beaming sunshine; a perfect reflection of how I felt - a fresh young inexperienced graduate student excited about a new opportunity that brought me to the Golden State. It was a summer of firsts for me: my first time in California, my first prolonged internship, my first project that generated positive meaningful results, my first attendance at a scientific conference, my first poster presentation, and even my first dip in the pacific ocean! I rushed into the lab that day, late of course because as a New Yorker I was oblivious to the fact that missing a train means at least a 20 minute wait for the next one!
I clearly remember my first day at the SENS Foundation research center in Mountain View, California. It was a gorgeous morning with clear skies and beaming sunshine; a perfect reflection of how I felt - a fresh young inexperienced graduate student excited about a new opportunity that brought me to the Golden State. It was a summer of firsts for me: my first time in California, my first prolonged internship, my first project that generated positive meaningful results, my first attendance at a scientific conference, my first poster presentation, and even my first dip in the pacific ocean! I rushed into the lab that day, late of course because as a New Yorker I was oblivious to the fact that missing a train means at least a 20 minute wait for the next one!
I went inside to find everybody hanging out in the tiny cafeteria room. One thing I quickly learned about SENS was the lack of a formal hierarchy; where an intern can share jokes with the CEO, enjoy a drink with the CSO, borrow a bike from the manager, get a comforting hug from her boss, and share a vegetarian meal with her supervisor. Matthew O’Conner (aka Oki), the head of MitoSENS took me into his office and sat down on his exercise ball while I got the luxury of taking his desk chair. From then on, I got acquainted to the idea of walking into his office and finding him bouncing up and down on his ball - I nicknamed him Mr. Bouncy which he corrected to Dr. Bouncy of course. I had the privilege of working with him and Gayathri Swaminathan, both very talented scientists. I was lucky enough to share an office with Gayathri, during which time I learned that she is not only brilliant, but humble, inspiring, and extremely patient (with my multitude of questions)!
The current project for mitoSENS is allotopic expression, which involves copying the mitochondrial DNA into the nucleus. My project required checking for integration of the DNA transfected into cells, and detecting RNA expression levels. By the end of the summer, I had done this successfully for 4 out of the 13 genes involved in oxidative phosphorylation that are still encoded by mitochondrial DNA. I spent my summer mostly doing PCRs (polymerase chain reaction), DNA and RNA isolations, cell culturing, and gel electrophoresis. I learned to perfect these techniques, to think critically when my results weren’t as expected, and to design experiments. My experience at SENS helped shape me into a more confident and better experienced scientist. I would definitely recommend volunteering for this foundation; the experience was educational, the research is open-minded, determined, and bold, and the staff is friendly, welcoming, and helpful.
I was also fortunate enough to have the chance to attend the SENS 5 conference in Cambridge at the end of my internship. This was a remarkable experience for me; I was exposed to an incredible amount of information on research in the aging field, and met fascinating people who are ambitious about ending aging. I am back in graduate school in New York now and I must say I am much better prepared to take on my thesis project. I’d like to thank everybody at the SENS foundation for their contribution towards making this experience so valuable for me.

View the full article
ImmInst
15 Oct 2011
To develop an unbreachable defense against cancer, SENS Foundation is pursuing the WILT (Wholebody Interdiction of Lengthening of Telomeres, or OncoSENS) strategy of preemptively deleting genes essential to the cellular telomere-maintenance mechanisms (TMM) from all somatic cells, while ensuring ongoing tissue repair and maintenance through periodic re-seeding of somatic stem-cell pools with autologous TMM-deficient cells whose telomeres have been lengthened ex vivo. Without a mechanism for extending its telomeres, the replicative potential of a cancer cell faces an absolute barrier to achieving clinically significant growth, and what is today a death sentence (or at best a chronic or potentially relapsing disease) is transformed into a benign lesion, of no more clinical significance than a cyst or plantar hyperkeratosis.(12-14)
At its core, then, WILT entails the ablation of some gene encoding an element of the telomerase holoenzyme. The strongest challenge to this approach, granting the periodic replenishment of somatic stem-cell pools with autologous but OncoSENS-ready stem cells, has been the possible existence of functions of TERT (telomerase reverse transcriptase -- the catalytic subunit of telomerase), other than the lengthening of telomeres itself. In recent years, several reports(1-10) have emerged claiming to have uncovered such functions, but generally in vitro and in nearly all cases under unphysiologically high and/or persistent forced expression of the telomerase gene. While it is not necessarily the case that TERT itself be knocked out -- ablation of the gene encoding the RNA template for TERT's reverse transcriptase is the other main candidate -- WILT is a sufficiently, complex, sweeping, and essential as the sole strategy currently proposed to render the human body ultimately immune to malignant disease that having one of our main options for implementation taken off of the table because of gene pleiotropy would be an unwelcome restriction on therapeutic options.
A direct in vivo test for deleterious effects of the deletion of TERT that do not involve the critical shortening of telomeres per se is somewhat challenging. Even first-generation TERT-/- mice do exhibit some mild shortening of the normal lifespan,(15) but such animals suffer some loss of tissue renewal across their lives. But a careful test of the development of TERT-/- mice would be expected to provide strong evidence on the subject one way or the other, provided that telomere lengths did not shorten excessively.
Nobel laureate Dr. Carol W. Greider, whose career in telomerase research and path to the Nobel prize began when she first identified the enzyme in 1984, has finally carried out such a test, and she and her collaborators have generated results that strongly support the safety of this element of WILT.
Seeking -- and Not Finding
To rigorously test for any non-TMM effects of TERT requires confidence that any phenotypic abnormalities are not related to telomere shortening per se. Greider's group built two strong safeguards against this major confounder. The first was the comparison of mice with TERT knockout and haploinsufficiency, to mice haploinsufficient and deficient in mTR, the murine TERC. Such mice have a fully functional catalytic telomerase subunits, but suffer progressive telomere shortening. They also made a novel selection for their background strain. In previous studies, TERT-/- mice have been generated on either a C57BL/6J or a 129/C57BL/6J mixed genetic background strain, both of which have "very heterogeneous and unusually long telomeres," again raising the possibility that any developmental abnormalities might be due to subpopulations of stem cells with critically-short telomeres. To avoid this problem, they instead performed their experiments in the CAST/EiJ mouse, a strain with telomere lengths similar to humans and homogeneous telomere length distributions.(11)
Telomere shortening and progressive impairments of tissue renewal in mTERT–/– and mTERT+/– mice on this background was similar to that in mTR–/– and mTR+/– mice, respectively. CAST/EiJ mTR–/– mice exhibit significant deficits in tissue renewal during adulthood, even in the first generation, and progressive worsening of the phenotype with successive generations, similar to what is seen in other background strains with these mutations and in the genetic anticipation observed in successive generations of families with autosomal dominant forms of the human genetic disorder of telomerase components, dyskeratosis congenita. Importantly, however, "mTERTâ»/â» mice, from heterozygous mTERTâº/â» mouse crosses, were born at the expected Mendelian ratio (26.5%; n = 1,080 pups), indicating no embryonic lethality of this genotype ... [and] show no additional phenotypes not seen in mTR–/– mice"(11) -- and even more importantly, "mTERT–/– mice show no additional phenotypes not seen in mTR–/– mice."(11)
Still Wingless
Amongst the studies claiming to have found evidence of a TMM-independent function of TERT, two(9.10) have drawn a putative connection to Wnt signaling. But in investigator-blinded comparisons of tissue sections from each group of mice, no evidence of defects related to loss of Wnt signaling were found in embryonic or adult pulmonary, renal, cerebral, or skeletal tissues. In particular, (9) had reported that some of their mTERT–/– mice were missing ribs, which was interpreted as a developmental defect of impaired Wnt signaling; comparison of wild-type and mTERT–/– CAST/EiJ and C57BL/6J mice showed normal numbers and morphology of ribs in all animals.(11) To test for possible developmental compensation for more subtle Wnt defects, they examined haploinsufficient crosses of CAST/EiJ mTERT+/– mice; none of the multiple defects observed in mice with knockouts of multiple different Wnt subtypes; again, no such phenotypes were observed.(11) To more directly test for defects in Wnt signaling, CAST/EiJ and C57BL/6J WT and mTERT–/– embryonic fibroblasts were transfected with a luciferase reporter plasmid with a Wnt3a ligand; luciferase expression was indistinguishable between WT and mTERT–/– cells.(11)
No Hairy Deal, No Silent Partner
Similarly, another group had reported excessive hair growth in mice with genetically augmented TERT activity under an unphysiologic promoter,(5) suggesting a possible role of TERT in follicular (and possibly stem) cell growth. But mTERT-/- mice exhibited no defects in hair loss.(11) Yet another group had suggested, based on unphysiologic cell models, that TERT might be involved in production of some small interfering RNAs as part of a ribonucleoprotein complex with RNA component of mitochondrial RNA processing endoribonuclease (RMRP);(8) again, no phenotypes suggestive of RMRP deficiency were observed in mTERT–/– mice.(11)
Opening the Lanes for A Long Drive Home
This little-heralded, meticulous investigation into the effects of ablation of the telomerase catalytic subunit in mice with human-like telomeres provides us with strong reassurance that, should it prove to be the preferred approach for implementing the OncoSENS strategy, the effects of knocking out TERT would be limited to those dictated by the loss of telomere-lengthening per se, and would not lead to an unintentional loss of some essential but hitherto-unknown phsyiological function. In light of the importance of WILT as an element of a comprehensive suite of rejuvenation biotechnologies,this news is welcome -- but insufficient, despite its rigor. SENS Foundation is funding additional research to further open up the path toward its realization. Amongst these are a research project in the lab of Dr. Jan Vijg, Chair of the Department of Genetics at Albert Einstein College of Medicine, who with research associate Dr. Silvia Gravina is performing the first single-cell analysis of the rate of incidence of epimutations in the aging mouse brain, to test for any unexpected involvement of non-cancerous mutations in the degenerative aging process, whose existence might render WILT an insufficient strategy to address nuclear genomic epimutations during the course of the "normal" life expectancies of today. Another is to monitor the effects of transplanting telomerase-deficient but ex vivo telomere-extended bone marrow into late-generation, TMM-disabled mice, so as to be certain that the niche of such animals (or, by implication, aging humans) will support the homing, engraftment, and initial development and differentiation of such cells; the necessary research is underway now thanks to a SENS Foundation grant to Dr. Zhenyu Ju of the Institute of Laboratory Animal Sciences and Max-Planck-Partner-Group on Stem Cell Aging in the Chinese Academy of Medical Sciences, and research partner of prominent telomere biologist Dr. K. Lenhard Rudolph.
WILT is the most ambitious plank in the SENS platform, with the longest and most logistically intricate path to development. The results of this and additional SENS Foundation-funded studies provide researchers with a confident basis to proceed toward an end to the scourge of cancer.
References
1. Ju Z, Jiang H, Jaworski M, Rathinam C, Gompf A, Klein C, Trumpp A, Rudolph KL. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat Med. 2007 Jun;13(6):742-7. Epub 2007 May 7. PubMed PMID: 17486088.
2. Flores I, Cayuela ML, Blasco MA. Effects of telomerase and telomere length on epidermal stem cell behavior. Science. 2005 Aug 19;309(5738):1253-6. Epub 2005 Jul 21. PMID: 16037417 [PubMed - indexed for MEDLINE]
3. Liu L, DiGirolamo CM, Navarro PA, Blasco MA, Keefe DL. Telomerase deficiency impairs differentiation of mesenchymal stem cells. Exp Cell Res. 2004 Mar 10;294(1):1-8. PMID: 14980495 [PubMed - indexed for MEDLINE]
4. Passos JF, Saretzki G, von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 2007;35(22):7505-13. PMID: 17986462
5. Sarin KY, Cheung P, Gilison D, Lee E, Tennen RI, Wang E, Artandi MK, Oro AE, Artandi SE. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature. 2005 Aug 18;436(7053):1048-52. PMID: 16107853 [PubMed - indexed for MEDLINE]
6: Masutomi K, Possemato R, Wong JM, Currier JL, Tothova Z, Manola JB, Ganesan S, Lansdorp PM, Collins K, Hahn WC. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci U S A. 2005 Jun 7;102(23):8222-7. Epub 2005 May 31. PubMed PMID: 15928077; PubMed Central PMCID: PMC1149439.
7: Geserick C, Tejera A, González-Suárez E, Klatt P, Blasco MA. Expression of mTert in primary murine cells links the growth-promoting effects of telomerase to transforming growth factor-beta signaling. Oncogene. 2006 Jul 20;25(31):4310-9. Epub 2006 Feb 27. PubMed PMID: 16501597.
8: Maida Y, Yasukawa M, Furuuchi M, Lassmann T, Possemato R, Okamoto N, Kasim V, Hayashizaki Y, Hahn WC, Masutomi K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature. 2009 Sep 10;461(7261):230-5. Epub 2009 Aug 23. PubMed PMID: 19701182; PubMed Central PMCID: PMC2755635.
9: Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H, McLaughlin M, Veenstra TD, Nusse R, McCrea PD, Artandi SE. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature. 2009 Jul 2;460(7251):66-72. PubMed PMID: 19571879.
10: Choi J, Southworth LK, Sarin KY, Venteicher AS, Ma W, Chang W, Cheung P, Jun S, Artandi MK, Shah N, Kim SK, Artandi SE. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008 Jan;4(1):e10. Epub 2007 Dec 13. PubMed PMID: 18208333; PubMed Central PMCID: PMC2211538.
11: Strong MA, Vidal-Cardenas SL, Karim B, Yu H, Guo N, Greider CW. Phenotypes in mTERTâº/â» and mTERTâ»/â» mice are due to short telomeres, not telomere-independent functions of telomerase reverse transcriptase. Mol Cell Biol. 2011 Jun;31(12):2369-79. Epub 2011 Apr 4. PubMed PMID: 21464209; PubMed Central PMCID: PMC3133422.
12: de Grey AD, Campbell FC, Dokal I, Fairbairn LJ, Graham GJ, Jahoda CAB, Porter ACG. Total deletion of in vivo telomere elongation capacity: an ambitious but possibly ultimate cure for all age-related human cancers. Ann N Y Acad Sci. 2004 Jun;1019:147-70. PubMed: 15247008.
13: de Grey AD. Whole-body interdiction of lengthening of telomeres: a proposal for cancer prevention. Front Biosci 2005;10:2420-2429. PubMed: 15970505.
14: de Grey AD. WILT: Necessity, feasibility, affordability. In: Fahy GM, West M, Coles LS, Harris SB (eds) The Future of Aging: Pathways to Human Life Extension. 2010; Springer, 667-684.
15: García-Cao I, García-Cao M, Tomás-Loba A, Martín-Caballero J, Flores JM, Klatt P, Blasco MA, Serrano M. Increased p53 activity does not accelerate telomere-driven ageing. EMBO Rep. 2006 May;7(5):546-52. Epub 2006 Mar 31. PubMed PMID: 16582880; PubMed Central PMCID: PMC1479549.
View the full article
ImmInst
24 Oct 2011
Accumulation of soluble and insoluble aggregates of beta-amyloid protein (Aß) and other malformed proteins accumulate in brain aging and neurodegenerative disease, leading progressively to neuronal dysfunction and/or loss. These have long been widely accepted to be drivers of Alzheimer's disease (AD) and other age-related dementias and neurological disorders such as Parkinson's disease, and it has recently become increasingly clear that neuronal protein aggregates are the main driver of "normal" cognitive aging. To prevent and reverse the course of neurodegenerative disease and age-related cognitive dysfunction, the regenerative engineering solution is therapeutic clearance of extracellular aggregates (such as Aß plaques) and intracellular aggregates (such as soluble, oligomeric Aß).
Immunotherapeutic Aß clearance from the brain is a very active field of Alzheimer's research, with at least seven passive, and several second-generation active, Aß vaccines currently in human clinical trials.(1) Of all Aß immunotherapies, he furthest advanced along the clinical pipeline are the passive monoclonal antibody vaccines bapineuzumab/AAB-001 (Janssen/Elan/Pfizer) and solanezumab/LY2062430 (Eli Lilly), both of which are currently in Phase III clinical trials. Other approaches, still in preclinical development, include the use of beta-amyloid-targeting affibodies, DNA and peptide vaccination targeting beta-amyloid epitopes, and catalytic cleavage of the beta-amyloid peptide itself. We now have a published report of preliminary findings from the first Phase I trial in an Aß-targeting vaccine with novel properties, and with the benefit of preliminary findings of outcomes that have only emerged with the experience of its forerunners in previous clinical trials.
Novel Target Antigen
The new contender -- Hoffmann-La Roche/Morphosys candidate gantenerumab (R1450 or RO4909832) -- is the first fully human anti-Aβ monoclonal antibody to enter clinical development: previous candidates have been humanized versions of murine antibodies, or derived from antibody fragments, or antibodies already present in pooled human immunoglobulin for injection (IVIgG). Gantenerumab was selected from a human phage display library and "optimized in vitro for binding with sub-nanomolar affinity to a conformational epitope expressed on amyloid-β fibrils ... In peptide maps, both N-terminal and central portions of Aβ were recognized by gantenerumab."(2) This, too, is a novel charcteristic of the new passive vaccine: the investigators claim it is unique amongst therapeutic antibodies, and certainly most anti-Aβ antibodies in clinical development recognize B-cell epitopes located either at the N-terminus of the protein (as does bapineuzumab) or a central span (eg. solanezumab); those that exploit T-cell targeted epitopes (including those elicited through active vaccination) map primarily to the central span and C-terminus of the peptide.(1)
Novel Clearance Mechanism
Gantenerumab may also be distinct from its passive vaccine competitors in using a cell-mediated mechanism of action for the removal of Aβ from the brain. AN1792, the first Aβ-targeting vaccine to enter clinical trials, was an active vaccine, and were mediated through T-cell responses specific to the carboxy terminal of the peptide; to date, passive antibodies have appeared to work by inducing efflux of Aβ from the brain, either through passage of the antibody through the blood-brain barrier (BBB) followed by sequestering of brain Aβ and then cotransportation of IgG-Aβ immune complexes back through the BBB (eg bapineuzumab), or through the "peripheral sink" mechanism of binding systemic Aβ and eliciting drawdown of soluble Aβ from behind it (eg. solanezumab). While still in preclinical development, the approach that SENS Foundation finds most exciting (on first principles, and based on results to date) is catalytic clearance of the beta-amyloid peptide itself.*
In contrast to all of these, gantenerumab, appears to be the first passive vaccine to stimulate microglial phagocytosis of Aβ. "In functional assays gantenerumab induced cellular phagocytosis of human amyloid-β deposits in AD brain slices when co-cultured with primary human macrophages";(2) "In ex vivo studies of human brain slices from an independent sample of patients who had AD ... Gantenerumab induced phagocytosis of human amyloid in a dose-dependent manner ex vivo."(3) Promisingly, and consistent with such a mechanism, when tested model AD mice bearing transgenic familial AD mutations in amyloid precursor protein and presenillin-2, "gantenerumab showed sustained binding to cerebral amyloid-β and, upon chronic [5 mo] treatment, significantly reduced small amyloid-β plaques by recruiting microglia and prevented new plaque formation. Unlike other Aβ antibodies, gantenerumab did not alter plasma Aβ, suggesting undisturbed systemic clearance of soluble Aβ."(2)
Those findings have now been supplemented with the first data derived from human clinical trials.
Phase I Clinical Trial Data
In a Phase I clinical trial,(3) 18 mild to moderate AD patients, aged 50-90 y, recruited from 3 university medical centers, were randomized to receive 7 monthly intravenous infusions of placebo or gantenerumab (60 or 200 mg). These doses were derived from previous, unpublished, single- and multiple-dose Phase I studies. In practice, however, while all subjects receiving the lower dose of antibody received the full course of therapy, few patients in the higher-dose group received all 7 infusions, due to early termination (see below): "One patient received 2 infusions, 2 patients received 3 infusions, 2 patients received 4 infusions, and 1 patient received 5 infusions."(3) Sixteen of these patients underwent carbon 11–labeled positron emission tomographic imaging with the Aβ-binding imaging agent, Pittsburgh Compound B (PiB-PET).
The results: based on PiB-PET, "The mean (95% CI) percent change from baseline difference relative to placebo (n = 4) in cortical brain amyloid level was -15.6% (95% CI, -42.7 to 11.6) for the 60-mg group (n = 6) and -35.7% (95% CI, -63.5 to -7.9) for the 200-mg group (n = 6)."(3) (See Figures 3 and 4, reproduced from original report, below). The approximate doubling of brain Aβ clearance in the higher-dose group is all the more remarkable considering the truncated course of therapy experienced by most (see below), and appears on a nominal basis to represent a faster rate of clearance than that previously reported for bapineuzumab over the course of 18 months (and that, without clear evidence of a dose-proportionate response).(4)
Reproduced from (4). Copyright American Academy of Neurology and the Authors.
The reason for the early termination of the trial was -- unfortunately but not unexpectedly -- the appearance of an adverse reaction in the high-dose gantenerumab group: MRI revealed that 2 subjects underwent transient periods of focal cerebrovascular inflammation or vasogenic oedema, coinciding with sites with the greatest local clearance of Aβ deposits.(3) Their appearance, while not welcome, would have been anticipated, based on their earlier occurrence in Phase II trials with bapineuzumab(4) and possibly solanezumab,(5) but ongoing study of these "amyloid-related imaging abnomralities" (ARIA)(11) now suggests that they are less concerning than they had initially appeared. Data from cerebral imaging studies of the general, non-AD population of Rotterdam show that haemosiderin abnormalities (ARIA-H) consistent with cerebral microbleeds occur in 3-15% of older adults,(6) and several studies presented at the 2011 Alzheimer’s Association International Conference (AAIC, formerly ICAD) suggest that vasogenic oedema (ARIA-E) may also be common in untreated subjects with the disease. In fact, reports there and elsewhere (including preclinical studies on the mechanism of such abnormalities, and a central blinded review of sequential brain images from the Phase II bapineuzumab trial) suggest that their increased occurrence in patients treated with Aβ-binding therapeutic antibodies is not only relatively mild and transient, but a potentially positive rough indicator of successful mobilization of brain Aβ.*
The combination of initial, dose-dependent reduction of brain Aβ as detected on PiB-PET, plus (ironically) the occurrence of vasogenic oedema at locations adjacent to the sites where clearance is greatest, is tantalizing evidence that gantenerumab is efficacious in removing these malformed proteins from the brain. The study was too small and early-phase to permit assessment of cognitive outcomes, but with the success of the Phase I trial, a Phase IIb clinical trial is now underway, which will test the effects of subcutaneous gantenerumab (225 or 105 mg every 4 weeks for 104 weeks) vs. placebo on cognitive and functional outcomes in subjects with prodromal AD (identified based on partner-observed, gradual reductions in memory and cerebrospinal fluid biomarkers, but without dementia (Mini-Mental State Exam score ≥24).
The Race is On
With promising preliminary human data and what appears to be a mechanism of Aβ clearance, the advancement of gantenerumab into preliminary efficacy testing places one more damage-removal therapeutic into the race for disease-modifying therapies for AD. Should it ultimately be successful and its migroglial cell-mediated mechanism of action be validated, it would have the advantage of being potentially in synergy with lysosomal fortification with novel hydrolases to enhance microglial lysosomal hydrolysis of engulfed Aβ.
Rejuvenation biotechnology is the application of the principles of regenerative medicine to the damage to cellular and molecular structures that accumulate in aging tissues -- the structural damage that disables those structures' function and leads to loss of homeostasis and the progressive rise in frailty, disease, disability, and death that people now suffer with age. Because the damage is multifarious, a platform of rejuvenation biotechnologies, rather than a single, all-encompassing "youth pill," will be required to achieve the robust rejuvenation of aging humans, restoring youthful health and vitality.
SENS is a strategy for engineering negligible senescence, based on this heuristic -- not a prescriptive list of therapies in development whereby it shall be executed. The rapidly-expanding group of agents, each with a meaningfully-distinct mechanism of Aβ clearance, entering into the clinical pipeline and in increasingly advanced stages of human clinical testing for the arrest and reversal of Alzheimer's disease, bodes well for the early achievement of the first rejuvenation biotechnology. SENS Foundation is proud to be engaged in its mission of catalyzing the progress toward a mature rejuvenation biotechnology industry; with that proof of concept, the wider biomedical field should become more alive to the application of the damage-removal heuristic to the many different kinds of aging damage underlying age-related disease. The engagement of a wide range of scientists in academia and industry, and aggressive funding of rejuvenation research, will accelerate progress toward a comprehensive panel of rejuvenation biotechnologies, and the achievement of thoroughgoing biomedical restoration of youthful health, vigor, and longevity.
* The theoretical advantages of catalytic antibodies as a mechanism for the therapeutic clearance of malformed protein deposits, combined with promising results to date in preclinical studies using such antibodies to clear brain Aβ, were key considerations leading to SENS Foundation's funding of research into catalytic antibodies for the removal of senile cardiac amyloidosis.
References
1: Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. Review. Erratum in: Nat Rev Neurol. 2010 Apr;6(4):183. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
2: Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, Messer J, Oroszlan K, Rauchenberger R, Richter WF, Rothe C, Urban M, Bardroff M, Winter M, Nordstedt C, Loetscher H. Gantenerumab: A Novel Human Anti-Aβ Antibody Demonstrates Sustained Cerebral Amyloid-β Binding and Elicits Cell-Mediated Removal of Human Amyloid-β. J Alzheimers Dis. 2011 Sep 28. [Epub ahead of print] PubMed PMID: 21955818.
3 : Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu ZX, Loetscher H, Santarelli L. Mechanism of Amyloid Removal in Patients With Alzheimer Disease Treated With Gantenerumab. Arch Neurol. 2011 Oct 10. [Epub ahead of print] PubMed PMID: 21987394.
4 : Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, Rodriguez Martinez de Liano S, Liu E, Koller M, Gregg KM, Schenk D, Black R, Grundman M. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010 Apr;9(4):363-72. Epub 2010 Feb 26. PubMed PMID: 20189881.
5: Bonetta L. Paris: Renamed ARIA, Vasogenic Edema Common to Anti-Amyloid Therapy. Alzforum (online resource). 2011 Jul 29. Accessed 2011-09-28.
6: Poels MM, Ikram MA, van der Lugt A, Hofman A, Krestin GP, Breteler MM, Vernooij MW. Incidence of cerebral microbleeds in the general population: the Rotterdam Scan Study. Stroke. 2011 Mar;42(3):656-61. Epub 2011 Feb 9. PubMed PMID: 21307170.
7: Zago W, Kinney G, Schroeter S, Khan K, Games D. Microvascular changes associated with passive immunotherapy in PDAPP mice - Potential implication for the etiology of vasogenic edema. Alzheimer’s Association International Conference, Paris. 2011 Jul 16-21. Abstract P3-052.
8: Sperling R, et al Revised estimates of incidence and risk factors for amyloid related imaging abnormalities (ARIA) in the phase 2 studies of bapineuzumab for mild to moderate Alzheimer's disease. Alzheimer’s Association International Conference, Paris. 2011 Jul 16-21. Abstract P4-438.
9: Salloway S, Sperling R, Honig, L Arrighi M, Wei H-L, Yuen E, Liu E, Morris K, Grundman M, Brashear R. Long-term follow-up of AD patients treated with bapineuzumab in phase 2. Alzheimer’s Association International Conference, Paris. 2011 Jul 16-21. Abstract O4-08-07.
10: Salloway S. Vasogenic edema in immunotherapy: Sign of efficacy or danger? Alzheimer’s Association International Conference, Paris. 2011 Jul 16-21. Abstract S5-01-05
11: Sperling RA, Jack CR Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011 Jul;7(4):367-85. PubMed PMID: 21784348.
View the full article
ImmInst
07 Nov 2011
Aging bodies become increasingly burdened over time with dysfunctional cells resistant to apoptotic or other clearance. The most well-known of these are so-called "senescent" cells, originally characterized by Leonard Hayflick as mitotic cells that reached growth arrest after a limited replicative lifespan (later associated with telomere attrition) under unphysiological conditions in culture. Later research has revealed that few cells reach a "senescent" state through sheer replicative exhaustion: instead, senescence has emerged as a programmed response to DNA damage or oncogenic stress, and as part of the resolution of wound healing.(1) Unfortunately, the near-term benefits of these functions -- in preventing damaged cells from progressing to cancer, and in preventing fibrosis --are coupled to deleterious long-term consequences, whose effects worsen as the burden of such cells rises with aging. First, the loss of mitotic competence of stem cells denies proliferative tissues of the capacity for renewal. Secondly, the secretory and other phenotypes of such cells progressively derange local and systemic metabolism and tissue function, rendering tissues more vulnerable to metastasis, promoting systemic inflammation, and otherwise impairing tissue function.(1-4)
To bypass the disruptive effects of the age-related accumulation of senescent cells, some investigators are working on possible ways to manipulate the signaling pathways involved in enforcing the senescent phenotype. This approach bears with it great risks, however, because of the very purposes of senescence to which allusion was made above: returning senescent cells to their normal differentiated function and replicative capacity could lead cells bearing oncogenic mutations to progress into metastatic disease, and aberrant resumption of the wound-healing response leading to fibrosis.(1,4) The regenerative engineering solution to this dilemma is therefore the ablation of such cells, to eliminate their contribution to age-related loss of homeostasis without reactivating the more acute risks against which the senescence machinery was activated in the first place.(5)
As widely covered in the mainstream press, a successful proof-of-principle study for this rejuvenation biotechnology has now been performed.(6)
The study was performed using several founder lines, bred onto a background strain of mice hypomorphic for BubR1 (BubR1H/H), a key component of the mitotic checkpoint machinery. Principal investigator Jan van Deursen, Professor of Biochem/Molecular Biology and of Pediatrics at the Mayo Clinic location in Minnesota, had already discovered(7) that BubR1H/H mice "have a markedly shortened lifespan and exhibit a variety of age-related phenotypes, including infertility, lordokyphosis, sarcopenia, cataracts, [subcutaneous] fat loss, cardiac arrhythmias, arterial wall stiffening, impaired wound healing and dermal thinning."(6) Some, but not all, of these phenotypes were associated with a high age-related incidence in senescent (p16Ink4a-positive) cells.(6-8) and van Deursen and colleagues had already demonstrated that breeding BubR1H/H mice onto a p16Ink4a homozygous-null genetic background attenuated their development of p16Ink4a-senescent cell-associated aging phenotypes and modestly increased their very low survivorship.(8) Imputation of these results specifically to the animals' age-related, low-BubR1-driven rise in p16Ink4a-expressing senescent cells was, however limited, both by the very nature of so-called "accelerated aging" models such as BubR1H/H(9) and by the lifelong, global absence of p16Ink4a expression in the backcrossed mice.
Seeds of Destruction and Renewal
To impute aging phenotypes directly to p16Ink4a-expressing senescent cells, van Deursen and colleagues with expertise in the aging and senescence of the relevant tissues developed and tested the effects of a pharmacologically-inducible system for the ablation of p16Ink4a-expressing cells. To create this system, investigators modified an approach used in earlier research, in which mice were bred with a variant on the Gene-Directed Enzyme Prodrug Therapy (or "suicide gene") paradigm,(11) using a drug (AP20187) that activated fusion protein apoptosis machinery in cells in which the macrophage- and adipocyte-specific minimal Fabp4 promoter was transcriptionally active.(10) To generate mice in which p16Ink4a-expressing cells could be similarly selectively ablated, van Deursen's team substituted a fragment of the p16Ink4a gene promoter for the Fabp4 promoter, thereby generating BubR1H/H;INK-ATTAC mice.(6) In such mice, then, p16Ink4a would still be under normal physiological regulation, and still be induced in an abnormally high number of cells due to the mitotic checkpoint dysfunction caused by BubR1 hypomorphism, leading to the same abnormally-rapid accumulation of high burdens of p16Ink4a-expressing senescent cells -- but administration of AP20187 would induce apoptosis selectively in such cells, purging the animals' tissues of senescent cells while leaving non-senescent cells unscathed.
Testing the System
A range of in vitro and in vivo tests was used to rigorously confirm the selectivity and sensitivity of the system's activation in, and ablation of, p16Ink4a-positive senescent cells.(6) In early-aging (2-mo old) BubR1H/H;INK-ATTAC mice, but not young (3-wk-old) mice, transcripts of the system and of reporter green fluorescent protein "were significantly elevated in [subcutaneous] adipose tissue, skeletal muscle and eye, but not in tissues in which endogenous p16Ink4a is not induced, including liver and heart."(6) Moreover, subcutaneous adipose of prematurely-aged transgenic mice exhibited high levels of staining for the senescence marker senescence-associated-β-galactosidase (SAβ-gal) and expressed high levels of several established markers of senescence, including p21, p19, interleukin-6, (insulin-like growth factor binding protein-2 (Igfbp2), and Pai-1; primary BubR1H/H;INK-ATTAC mouse embryonic fibroblasts forced artificially into senescence by oncogenic Ras or serial passage exhibited a subpopulation that was both GFP+ and stained positively SAβ-gal. BubR1H/H;INK-ATTAC muscle cells and lens did not stain for SAβ-gal, but did exhibit selective induction of the "senescence genes."(6)
When BubR1H/H;INK-ATTAC mouse bone marrow cells were pushed into senescence in vitro by the PPAR-γ-activating drug rosiglitazone, a subpopulation of the cells exhibited high levels of INK-ATTAC expression and GFP, coupled with SAβ-gal staining; subsequent to treatment with the INK-ATTAC activating drug, these cells rapidly entered into apoptosis, and within 48 h were either destroyed or in the cell death process.(6)
Ablation of Senescent Cells Retards Age-Related Tissue Degeneration
As a first approach, the investigators abrogated the premature age-related rise of p16Ink4a-senescent cell burden in the tissues of BubR1H/H;INK-ATTAC mice by initiating a lifelong course of senescent-cell-ablating AP20187 treatment at weaning. At 9-10 mo of age, such mice were then compared a to age-matched cohorts of untreated BubR1H/H;INK-ATTAC mice, and to BubR1H/H mice lacking the INK-ATTAC system for selective ablation of p16Ink4a-expressing cells. Relative to both control cohorts of the same age, 9-10 mo old treated mice exhibited dramatically more youthful tissues. Consistent with earlier results, their burden of p16Ink4a-positive senescent cells in muscle, eye, and adipose tissues were far lower. Their muscle fibers had larger diameters, their treadmill endurance was greater and they covered more distance on them. Treated mice had fewer cataracts, and less lordykyphosis. And they suffered less lipoatrophy, with larger fat deposits in multiple depots, higher individual adipocyte volumes, and more proliferating cells marked with BrDU (see Figure 1, below).(6) No treatment-related adverse events presented themselves.(6)
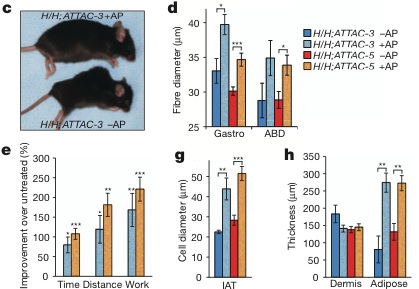 H/H;INK-ATTAC Mice" />
H/H;INK-ATTAC Mice" />
Figure 1: Amelioration of "Premature Aging" Phenotypes in Treated and Untreated BubR1H/H;INK-ATTAC Mice. AP=AP20187 treatment. Reproduced from (6). © Nature Publication Group.
Importantly, "premature aging" phenotypes observed in BubR1H/H mice over time, but that are in tissues where p16Ink4a-positive senescent cells do not accumulate with aging, were not alleviated by drug treatment. Thus, these animals exhibit premature cardiac arrhythmias and stiffening of the arterial wall, and cardiac failure appears to be the main cause of death; yet these tissues are not burdened with an abnormally-high burden of p16Ink4a-senescent cells, and accordingly, ablation p16Ink4a-positive senescent cells in these animals had little tissue-specific or survivorship phenotypic impact.(6)
Following this initial test of abrogating the early, age-related rise in p16Ink4a-expressing cell burden, the investigators probed the effects of leaving BubR1H/H;INK-ATTAC to undergo 5 months of rapid "premature aging" (and thus, to the attendant accumulation of high levels of p16Ink4a-positive cells and onset of "early-aging" phenotypes), and only then inducing ablation of senescent cells with the INK-ATTAC drug-activated system (see Figure 2 (g) below).(6) At that point, the animals' cataracts had already reached peak age-related severity, and remained stable after 5 months of further aging irrespective of treatment. But muscle fibers that continued to atrophy over the ensuing 5 months in control animals remained at their more youthful diameters in animals whose p16Ink4a-positive senescent cells had been ablated, and treadmill times, distance traveled, and work outputs were maintained at substantially more youthful levels (Figure 2, (a) and (b) below). Similarly, the degeneration of adipose tissue cells and depots that occurred over the course of the next 5 months in control animals was virtually abrogated, leaving 10 mo-old animals with substantially the same subcutaneous and other fat tissue (in depth and in cell volume) as they had enjoyed in their relative youth, when treatment was first initiated.(6)
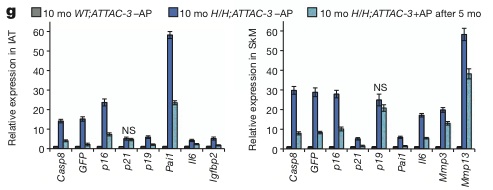
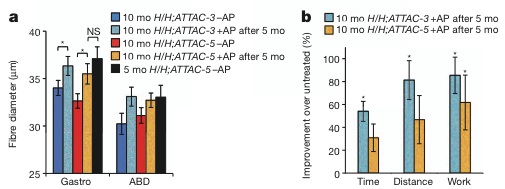
Figure 2: Ablation of Senescent p16Ink4a-Expressing Cells Maintains Youthful Muscle and Adipose Tissues. Reproduced from (6). © Nature Publication Group.
Rejuvenation Implications
As noted above, studies involving the use of putative "premature aging" models must be interpreted with caution, as the designation inevitably involve an element of petitio principii: from a subset of similar phenotypes are drawn conclusions of similar aetiology, and from this, further conclusions about the "normal" degenerative aging process (and its biomedical amelioration) are too-readily drawn before the thesis itself has first been established.(9) Indeed, the degenerative aging process is by definition one in which the organism progressively accumulates damage to its cellular and molecular components over time, so any genetic or environmental factor that leads to a greater burden of such damage will bear some resemblance to the aging phenotype, irrespective of the causal origin of the defect or its relationship to "normal" aging.
In the case of this new report,(6) however, while caution is still merited, the nature of the intervention used makes the study relatively free of such complications. The investigators did not simply modulate or normalize the very thing that the mutation (in this case, to the mitotic checkpoint component BubR1) itself disrupts, as in other widely-publicized studies involving putative "accelerated aging" (eg. (12,13)). Rather, the defective checkpoint system was left to proceed, and one of its downstream consequences, which was still under normal regulation -- and one known to be directly induced by the normal degenerative aging process -- was reversed at the structural level, by clearing out the p16Ink4a-positive senescent cells that had accumulated to an abnormal degree in their tissues. This left some aspects of the abnormal "progeroid" phenotype in these organisms (the cardiovascular defects) intact, but illustrated the dysfunctional consequences of having tissues riddles with such cells. While still of abnormal origin, there is no strong reason to think that the ongoing effects of a rising burden of such cells would not be similar -- and thus, that the effects of ablating such cells are uninformative about the effects of a similar intervention in "normally" aging bodies.*
The links to aging phenotypes, and their near-arrest by ablation of p16Ink4a-expressing senescent cells, appear to be dramatic illustrations of the deleterious effects of the age-related rise in the burden of senescent cells in genetically-intact mammals. The fact that it was the removal of such cells from aging tissues that arrested multiple aging phenotypes is of special importance to the rejuvenation biotechnology approach to preventing and reversing age-related disease and disability: it clearly identifies the damage itself, rather than the abnormal function of either p16Ink4a (which was under normal, physiological regulation, rather than being pharmacologically modulated, or knocked out as in their previous report(8)) or BubR1 (mutation of which, and its direct metabolic sequelae, was not affected by the intervention).
And there are reasons to believe that the resulting arrest of multiple aspects of tissue aging by removal of p16Ink4a-expressing senescent cells would indeed translate into the tissues of genetically-intact mice -- or humans.
Sarcopenia
The fact -- and disabling and fatal consequences -- of age-related decline in muscle quality and quantity is widely known, but the contribution of "senescent" cells to this degenerative process is not. While some studies have reported no decline in satellite cells (muscle progenitor cells) with aging, others (eg. (14,15)) have found age-related satellite cell attrition consistent with the senescence of a subset thereof; moreover, one such study (15) reported that decreases in the number and quality of satellite cells with aging are reliably associated with elevated expression of p16Ink4a (contrary to (14)), and with secretory and proteomic abnormalities consistent with a rising burden of senescent cells. Consistent with a causal relationship, (16) reports that the prevalence of limited physical functioning in aging varies depending on p16Ink4a allelic variation, consistent with variations in rate of stem cell attrition with senescence.
There is therefore good reason to expect that the profound arrest of sarcopenic phenotypes observed in p16Ink4a-senescent cell ablated BubR1H/H "premature aging" mice would translate into the human case.
Lipoatrophy
While less well-known (masked as it is and placed out of focus by the overall age-related body composition shift from lean mass to adiposity), there is none the less significant age-related subcutaneous lipoatrophy in aging, most visibly in the sunken appearance of the face. Part of this is a pathological redistribution of adipose from the subcutaneous to the visceral depot, but it now emerges that the subcutaneous depot becomes qualitative as well as quantitatively abnormal in the degenerative aging process also suffers genuine age-related lipoatrophy and lipodystrophy -- and that p16Ink4a-driven cellular senescence is at the heart of it.
Subcutaneous adipose tissue contributes to maintenance of insulin sensitivity and other aspects of metabolic homeostasis, through the production of adipose-specific endocrine factors such as adiponectin. Surgical removal of subcutaneous fat reduces adiponectin levels and insulin sensitivity, and transplantation of subcutaneous fat increases both.(17) Slow-aging growth hormone receptor knockout (GHRKO) mice are obese, but highly insulin sensitive: in such animals, surgical removal of visceral adipose tissue impairs insulin secretion and peripheral insulin action, in part by reducing adiponectin production. (21) Moreover, while the link between excessive visceral adipose tissue and age-independent diabetes and metabolic syndrome widely known, recent studies suggest instead that it is instead the accumulation of senescent subcutaneous adipocyte progenitors -- and their abnormal metabolic function -- that drives similar diabetes-like phenotypes during the "normal" aging process.(3,18; cf. 19,20) Even in visceral fat, it has recently emerged that the obesity-driven rise in inflammation and insulin resistance is associated with an abnormal accumulation of senescent cells, albeit senescent endothelial cells rather than adipocytes.(20) It was this emerging line of research that van Deursen's collaborator Dr. James Kirkland presented at the fifth annual Strategies for Engineered Negligible Senescence Conference (SENS5) in September of this year,(18) and it was his expertise in the senescence of adipose tissue that he contributed to the new report on the effects of ablating such cells.(6)
Again, then, there is significant evidence consistent with a role of cellular senescence in age-related lipodystrophy and lipoatrophy, and for the benefits observed in treated mice in these studies to translate into aging humans. It is unfortunate that the investigators did not assess insulin secretion, insulin action, or systemic inflammation in early-aging BubR1H/H;INK-ATTAC mice, with and without ablation of senescent adipose cells, but reasonable to be optimistic that doing so would yield some normalization of age-related metabolic abnormalities.
Cataract
There is only the most tentative of evidence suggesting a link between cellular senescence and cataract in "normal" aging.(22) Absence of evidence is, however, not evidence of absence, and certainly the inflammatory secretory profile of senescent cells would, if present, likely accelerate the degenerative course of the disease.
Cancer
A great deal of evidence has now been amassed that stromal cell senescence plays an important role in laying the groundwork for tumor metastasis, promoting cell proliferation with inflammatory cytokines, encouraging angiogenesis, and degrading the tumor-suppressive action of an intact extracellular matrix.(1) One clear disadvantage of using these "early-aging" mice is that they die to early to develop cancer -- too early for ablation of p16Ink4a-positive senescent cells to impact the course of the disease. It would indeed be of great interest to see whether ablation of stromal p16Ink4a-expressing senescent cells, in otherwise genetically intact INK-ATTAC animals without existing tumors, would be at lower risk for cancer, and put any tumors they might develop a less malevolent trajectory, than untreated mice -- although even such a study would likely show limited effect compared to what might be anticipated in a large mammal model, since even normally-aging mice rarely suffer metastatic disease to the extent of aging humans, as sheer primary tumor volume is generally sufficient to be fatal to mice.
Other Tissues
As the investigators note, the rapid age-related arterial stiffening and cardiac arrhythmias that appear to be at cause for the majority of deaths in BubR1H/H mice were not attenuated by ablating p16Ink4a-expressing senescent cells -- but these tissues had little burden of such cells, so this finding reinforces the conclusion that the multiple aging phenotypes arrested in these mice when senescent cells were ablated is attributable specifically to the removal of their baleful influence on local tissues. On the other hand, there are many other tissues -- notably, the kidney and articular cartilage -- where p16Ink4a-expressing senescent cells appear to be a contributing factor to human and murine degenerative aging, but which were not evaluated in treated or control mice in this study, and it would be of interest to see the effects of ablation of p16Ink4a-positive senescent cells.
Moreover, there are yet other cell types -- such as visceral adipose tissue macrophages and cytotoxic CD8+ T-cells -- in which the age-related supernumerary accumulation of dysfunctional and apoptosis-resistant cells appears to play a highly deleterious role on tissue function, but where the cells are not "senescent" cells in the classical sense of p16Ink4a expression and the senescence-associated secretory profile observed in senescent fibroblasts. This study (6) cannot provide evidence directly on the effects of ablating such cells, but it does provide an analogous proof-of-concept for the approach. SENS Foundation is funding ongoing work in the lab of Dr. Janko Nikolich-Zugich to investigate the effects of clearance of anergic, "senescent" cytotoxic CD8+ T-cells on immunosenescence,(22) and is interested in the targeting of other such cells.(2)
Arrest vs Reversal
In the new study, p16Ink4a-expressing senescent cells were ablated either at weaning or some months later, and assessed some months later. Remarkably enough, the removal of such cells arrested tissue degeneration, holding the muscles and adipose tissue (and, when administered before cataract was mature, lens opacification) at approximately the same relatively youthful condition prevalent when the inducing drug was first administered (see eg. Fig 2(a) above).(6) This is consistent with the deleterious effect of such cells on tissue function, and with the researchers' conclusion that “the observed improvements in skeletal muscle and fat of late-life treated 10-month-old BubR1H/H;INK-ATTAC-5 mice reflect attenuated progression of age-related declines rather than a reversal of ageing”.(6) However, it would be useful to see a more thorough analysis of the effect of ablating p16Ink4a-expressing senescent cells, and whether there may instead be evicence of a short-term rejuvenation of tissue function that is slowly lost over time to rising levels of other kinds of aging damage that INK-ATTAC activation does not address. Indeed, as as illustrated by the lack of effect of p16Ink4a-expressing cell ablation on lifespan, and by the ongoing degeneration of tissues (such as the heart) in which p16Ink4a-postive senescent cells are not a driver of "early aging," true rejuvenation requires a comprehensive suite of rejuvenation biotechnologies to remove all forms of aging damage from the aging body.
Translation for Human Rejuvenation Biotechnologies
The investigators boldly, but rightly, conclude that
Our proof-of-principle experiments demonstrate that therapeutic interventions to clear senescent cells or block their effects may represent an avenue for treating or delaying age-related diseases and improving healthy human lifespan.(6)
How might the results of this intervention be translated for human rejuvenation therapies?
There is already evidence that senescent cells are targeted by the innate immune system.(24-28) Dr. Judith Campisi, in fact, has found that activating NKG2D receptors on natural killer (NK) cells engage MHC class I chain-related protein A and B (MICA/B) ligands on senescent cells, leading to their NK-induced apopotsis and subsequent clearance.(29) MICA/B ligands are also used to activate tumor cell destruction by NK cells via NKG2G binding, and tummors evolve resistance by several mechanisms to reduce cell-surface MICA abundance;(30) however, the natural selection mechanisms that drive the evolution of such defenses do not apply to growth-arrested cells. Dr. Campisi has found instead that a minority of senescent cells evade destruction by secreting high levels of matrix metalloproteinases (MMPs), which cleave MICA/B ligands and preventing NKG2D binding.(29) This has led to the hypothesis that the great majority of such cells are destroyed over the lifetime by innate immunity, and that the specific senescent cells that do accumulate with aging are precisely those who had variants that allow MMP overexpression, in a kind of "one-off" very temporally-extended kind of selection process. Potentially, a kind of intervention that could overcome this resistance to endogenous clearance mechanisms would allow for the purgation of senescent cells from aging tissues.
SENS Foundation is currently funding work by Dr. Kevin Perrot in Campisi's laboratory, screening compounds for their effectiveness in eliminating cells exhibiting the classical senescence-associated secretory phenotype (SASP)(1) following senescence induced by treatment with 10 gray of ionizing radiation. To date, a screen of the collection of FDA-approved drugs in the Prestwick Library has identified 400 such agents which have demonstrated effectiveness at lowering secretion of IL-6, a component of SASP whose concentration tends to rise systemically with aging and here used as a preliminary marker of SASP as a phenotype. In particular, Dr. Perrot has recently identified some members of a class of compounds that lower the SASP in irradiated-senescent cells, without reversing growth arrest, and he is currently investigating the mechanisms of this phenomenon.
None of the compounds screened to date have exhibited selective toxicity against senescent (vs. non-irradiated, non-IL-6-secreting) cells. However, the library of compounds available for screening by Dr. Perrott is now expected to expanded substantially, thanks to a research partnership recently established between the Buck Institute and Biotica Technology Ltd to work with their specialized pipeline of novel polyketide compounds, potentially bridging the long-established focus of SENS Foundation and the Campisi lab at the Buck on the role of senescent cells in the degenerative aging process, with Biotica's focus on clinical therapeutics and the wound response and resolution process. Discussions for still other proprietary small-molecule libraries for screening are ongoing.
It is clear that there is substantial distance yet to be traveled. Multiple cell types acquire distinctive "senescent" phenotypes on a cell-type-specific basis, and will require ablation to achieve comprehensive rejuvenation. However, this important proof-of-principle from Dr. van Deursen's laboratory, and the key validation of the scope of the effects of ablation of these particular senescent cells facilitated by his collaboration with the LeBrasseur and Kirkland labs at Mayo, stands as a key landmark in moving toward the removal of their baleful influence on aging tissues. As ever, SENS Foundation is committed to making investments in critical-path research to advance this key but heretofore-neglected line of biomedical research out of the laboratory, into the clinic, and to uniting the multiple strands of rejuvenation biotechnologies into a comprehensive panel for the restoration of the health, vigor, and open futures of aging humanity.
*The principle caveat would be that the interaction of senescence with defective mitotic checkpoin function within such cells, and the effects upon their neighbors of their state and of their senescence-associated secretory phenotype, would very likely cause some phenomena that would not be observed in p16Ink4a-senescent cells or in their effects on neighbors.
References
1: Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011 Feb 21;192(4):547-56. Epub 2011 Feb 14. Review. PubMed PMID: 21321098; PubMed Central PMCID: PMC3044123.
2: Burton DG. Cellular senescence, ageing and disease. Age (Dordr). 2009 Mar;31(1):1-9. Epub 2008 Sep 4. PubMed PMID: 19234764; PubMed Central PMCID: PMC2645988.
3: Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, Kirkland JL. Fat tissue, aging, and cellular senescence. Aging Cell. 2010 Oct;9(5):S26(Abs 49). doi: 10.1111/j.1474-9726.2010.00608.x. Epub 2010 Aug 15. Review. PubMed PMID: 20701600; PubMed Central PMCID: PMC2941545.
4: Naesens M. Replicative senescence in kidney aging, renal disease, and renal transplantation. Discov Med. 2011 Jan;11(56):65-75. Review. PubMed PMID: 21276412.
5: de Grey AD. Foreseeable pharmaceutical repair of age-related extracellular damage. Curr Drug Targets. 2006 Nov;7(11):1469-77. Review. PMID: 17100587 [PubMed - indexed for MEDLINE]
6: Baker DJ, Wijshake T, Tchkonia T, Lebrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16(Ink4a)-positive senescent cells delays ageing-associated disorders. Nature. 2011 Nov 2. doi: 10.1038/nature10600. [Epub ahead of print] PubMed PMID: 22048312.
7: Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004 Jul;36(7):744-9. Epub 2004 Jun 20. PubMed PMID: 15208629.
8: Baker DJ, Perez-Terzic C, Jin F, Pitel K, Niederländer NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, Eberhardt NL, Terzic A, van Deursen JM. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008 Jul;10(7):825-36. Epub 2008 May 30. PubMed PMID: 18516091; PubMed Central PMCID: PMC2594014.
9: Miller RA. 'Accelerated aging': a primrose path to insight? Aging Cell. 2004 Apr;3(2):47-51. Review. PubMed PMID: 15038817.
10: Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005 Jul;11(7):797-803. Epub 2005 Jun 19. PubMed PMID: 15965483.
11: Both GW. Gene-directed enzyme prodrug therapy for cancer: a glimpse into the future? Discov Med. 2009 Oct;8(42):97-103. Review. PubMed PMID: 19833053.
12: Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011 Feb 17;470(7334):359-65. Epub 2011 Feb 9. Erratum in: Nature. 2011 Jul 14;475(7355):254. PubMed PMID: 21307849.
13: Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadiñanos J, Horner JW, Maratos-Flier E, Depinho RA. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011 Jan 6;469(7328):102-6. Epub 2010 Nov 28. PubMed PMID: 21113150; PubMed Central PMCID: PMC3057569.
14: Carlson ME, Suetta C, Conboy MJ, Aagaard P, Mackey A, Kjaer M, Conboy I. Molecular aging and rejuvenation of human muscle stem cells. EMBO Mol Med. 2009 Nov;1(8-9):381-91. PubMed PMID: 20049743; PubMed Central PMCID: PMC2875071.
15: G. Butler-Browne, M.-C. LeBihan, A. Bigot, D. Furling, F. Svinartchouk, D. Bechet, V. Mouly. Identification of biomarkers of human muscle aging and senescence. Rejuvenation Res. 2007 Sep;10(Suppl1):S22(Abs 14).
16: Melzer D, Frayling TM, Murray A, Hurst AJ, Harries LW, Song H, Khaw K, Luben R, Surtees PG, Bandinelli SS, Corsi AM, Ferrucci L, Guralnik JM, Wallace RB, Hattersley AT, Pharoah PD. A common variant of the p16(INK4a) genetic region is associated with physical function in older people. Mech Ageing Dev. 2007 Mar 27; [Epub ahead of print] PMID: 17459456 [PubMed - as supplied by publisher]
17: Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008 May;7(5):410-20. PubMed PMID: 18460332; PubMed Central PMCID: PMC3204870.
18: Kirkland JL. Aging, Adipose Tissue, and Cellular Senescence. Abstracts of Strategies for Engineered Negligible Senescence (SENS) Fifth Conference. August 31-September 4, 2011. Cambridge, United Kingdom. Rejuvenation Res. 2011 Aug;14 Suppl 1:S11-45. PubMed PMID: 21847798.
19: Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F, Komuro I. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009 Sep;15(9):1082-7. Epub 2009 Aug 30. PubMed PMID: 19718037.
20: Villaret A, Galitzky J, Decaunes P, Estève D, Marques MA, Sengenès C, Chiotasso P, Tchkonia T, Lafontan M, Kirkland JL, Bouloumié A. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes. 2010 Nov;59(11):2755-63. Epub 2010 Aug 16. PubMed PMID: 20713685; PubMed Central PMCID: PMC2963533.
21: Bartke A. Effects of Calorie restriction in long-lived mice. Presentation at CR VII, Seventh Calorie Restriction Society Conference, Las Vegas, NV, October 26-29 2011.
22: Zhang ZF, Zhang J, Hui YN, Zheng MH, Liu XP, Kador PF, Wang YS, Yao LB, Zhou J.Up-Regulation of NDRG2 in Senescent Lens Epithelial Cells Contributes to Age-Related Cataract in Human. 2011;6(10):e26102. Epub 2011 Oct 17. PubMed PMID: 22043305; PubMed Central PMCID: PMC3197158. doi:10.1371/journal.pone.0026102
23: Smithey MJ, Renkema KR, Rudd BD, Nikolich-Žugich J. Increased apoptosis, curtailed expansion and incomplete differentiation of CD8+ T cells combine to decrease clearance of L. monocytogenes in old mice. Eur J Immunol. 2011 May;41(5):1352-64. doi: 10.1002/eji.201041141. Epub 2011 Apr 14. PubMed PMID: 21469120.
24: Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-{kappa}B promotes senescence and enhances chemosensitivity. Genes Dev. 2011 Oct 15;25(20):2125-36. Epub 2011 Oct 6. PubMed PMID: 21979375.v
25: Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006 Feb;130(2):435-52. PubMed PMID: 16472598.
26 : Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008 Aug 22;134(4):657-67. PubMed PMID: 18724938; PubMed Central PMCID: PMC3073300.
27: Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007 Feb 8;445(7128):656-60. Epub 2007 Jan 24. Erratum in: Nature. 2011 May 26;473(7348):544. PubMed PMID: 17251933.
28: Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C, Petrucci MT, Guarini A, Foà R, Santoni A. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009 Apr 9;113(15):3503-11. Epub 2008 Dec 19. PubMed PMID: 19098271.
29: Campisi J. New tricks for old cells. Understanding Aging: Biomedical and Bioengineering Approaches. 2008 Jun 27-26, UCLA.Program p.4-5.
30: Fuertes MB, Girart MV, Molinero LL, Domaica CI, Rossi LE, Barrio MM, Mordoh J, Rabinovich GA, Zwirner NW. Intracellular retention of the NKG2D ligand MHC class I chain-related gene A in human melanomas confers immune privilege and prevents NK cell-mediated cytotoxicity. J Immunol. 2008 Apr 1;180(7):4606-14. PubMed PMID: 18354183
View the full article
ImmInst
03 Nov 2011
In this three part series, SENS Foundation researcher Max Peto describes the pathophysiology of age-related macular degeneration (ARMD), and how we direct our internal research efforts to reverse the pathology of this terrible, debilitating disease.
The first part of this series will discuss the macro-physiology of the eye, describing the major structural features of the eye. The second part will “zoom-in” to the biochemistry of the visual cycle and the pathophysiology of ARMD will be described. Finally the third part will include a discussion on how we are approaching the development of potential therapeutics internally at the SENS Foundation Research Center, and how these strategies relate to the pathophysiology of ARMD.
Part I – Macro-anatomy of the human eye
While first reading material associated with macular degeneration, I discovered that there are many parts of the eye, and these are necessarily small. An understanding of how these many small structures interact with one another and work together to enable vision is critical in understanding the pathophysiology of ARMD. However, there is an even greater level of detail involved in understanding the structure and function of the eye than one might first think. First, there are larger macrostructures, such as the macula, retina, choroid, and others. But then there are smaller microstructures, such as rod, cone, and RPE cells, and relationships among these are also critical to understanding ARMD. In this post, I’ll first describe the macrostructures (including pictures), and discuss how they are related to one another. In the next post (Part II), I’ll zoom into the microstructures and discuss the biochemical details of the pathophysiology of ARMD.
Macro-anatomy of the human eye
First, I’ll start off with a good picture:

I won’t focus on many of these parts of the eye, as they’re not particularly relevant to ARMD. But I now bring your attention to those which do. These are the:
-
Choroid (3),
-
Retina (7), and
-
Macula and fovea (8)
I’ll start by describing the macula (this is macular degeneration we’re talking about).
The macula is the darker-colored part of the above picture, labeled “8.”. The following is a useful picture of an actual macula of a right eye:

Note again how the macula (also called the “macula lutea” or the “macula of the retina”) is the dark spot in at the center of the retina in the back of the eye, and has a high density of photoreceptor cells, particularly in the “fovea” or “foveal region” of the macula. Note that the macula sits at the back of the eye, directly inward from the lens of the eye, enabling highly-focused central vision by specially-processing light that enters straight into the eye and travels directly to the back of the eye. From this information, one can see that the above picture is not quite back-to-front of the eye, but was taken obliquely.
The fovea is a notable part of the macula, which has the highest concentration and density of cone cells in the eye. The macula, and more specifically the fovea, enables high-acuity central vision.
The retina is large, thin structure, and is the shape of ~70% of a sphere that sits in the back of the eye on top of the choroid layer. I think of the retina as a spherical bowl or round-bottomed cylinder of nerve cells into which light is funneled. This can be better visualized by thinking about the first picture above, which is a cross-section of an illustrated eye. I’m referring to the place where the two layers are pulled away from the back of the eye, illustrating the retina resting on the choroid. The retinal nerve cells transmit light signals to the brain via the optic nerve.
The retina sits on the choroid layer, which is the vascularized layer providing a blood (and thus nutrient) supply to the eye. I will mention this again later, but keep in mind that the retinal pigment epithelial (RPE) cells sit almost directly on top of the choroid layer. This interplay between RPE cells, photoreceptor cells, and the choroid is going to be relevant in a later discussion on photoreceptor cell metabolism (particularly waste metabolism).
To review, from the back (deepest part) of the eye to the front:
-
The retina sits on the choroid layer, a layer consisting of blood vessels and capillaries which provide the eye with nourishment.
-
The retina is a spherical light-funnel which sits in the back of the eye on the choroid, and is composed of ten layers, including nerve tissue which transmits signals to the optic nerve which are later interpreted as vision in the brain.
-
The macular region is situated on the center of the retina in the back of the eye, directly inward from the lens where light is allowed into the eye.
-
The fovea is a special, central part of the macular region, and has a very high concentration of cone-type photoreceptor cells, which are critical in enabling high-acuity central vision (which is lost in ARMD).
At the RC, we frequently talk about retinal pigmented epithelial cells, also known as RPE cells. RPE cells are actually one layer of ten which make up the retina, so keep in mind that RPE cells are actually a small part of the retina (which is why they’re called retinal PE cells).
In the next post, I’ll elaborate on RPE cells, photoreceptor cell metabolism, the visual cycle, and how these interact to cause the pathophysiology of age-related macular degeneration.
View the full article
ImmInst
25 Nov 2011
Rejuvenation biotechnology encompasses a suite of advanced medical therapies, each aimed at the removal, repair, replacement, or rendering harmless of a form of cellular or molecular damage that accumulates in aging tissues over time, impairing their function. Through the comprehensive abatement of all such aging damage to levels approximating those of younger adults, tissue structure and function can be made more youthful, restoring the health and vigor of aging persons to that of persons years or decades younger. This approach is most prominently under pursuit in the development of cell therapy and tissue engineering, of which the most striking success to date has been the use of fetal and embryonic mesencephalic tissue grafts to replace dopaminergic [DA] neurons lost to the age-related neurodegenerative processes driving Parkinson's disease (PD).(0) Such transplants reinnervate and partly functionally integrate into the patient striatum, locally restoring missing DA release needed for fine motor control.
While the results of such grafts have often been striking temporary improvements in the major motion disorder symptoms in patients, significant improvements in the protocol are clearly required. The supply of fetal midbrain tissue for grafting is both inherently limited and subject to politically-imposed restrictions; as well, the grafts are significantly immunogenic, leading to rejection in some cases and necessitating immunosuppression in most patients. moreover, the degree of clinical response has been highly variable,moreover, the degree of clinical response has been highly variable, gains have proven impermanent, and ~15% of patients have developed substantial dyskinesias during the "off" phase of levodopa treatment.
Many of these difficulties are expected to be amenable to the replacement of the crude cell supplies used in these trials with pure dopaminergic neurons derived from pluripotent stem cells, such as embryonic stem cells (ESC), induced pluripotent stem cells (iPS), or somatic cell nuclear transfer (SCNT -- "theraputic cloning"). ESC-derived differentiated cells and progenitors appear to enjoy some degree of immunological privilege,(1) whereas iPS cells would be immunologically native and free of rejection risk, with the possible exception of mitochondrially-derived immunogenicity in SCNT. Moreover, the graft-induced dyskinesias now appear to be primarily the result of the presence of serotonergic neurons in the mixed cell population of the graft,(2) which would be eliminated by specific derivation of dopaminergic neurons from ESC or other pluripotent cells. And the nearly-unlimited replicative capacity of pluripotent cells, combined with an efficient and stable means of differentiation, could provide such cells in quantities needed for widespread clinical use in frank PD and earlier in brain aging.
In murine models of PD, DA neurons derived from mouse ESC have been found highly effective in reversing motor symptoms, but the performance of DA neurons derived from human pluripotent stem cells has so far been poor, and the stability of differentiation dubious. A new study exploits a novel differentiation strategy to resolve these difficulties, leading to robust engraftment of human ESC- and iPS-derived DA neurons and substantial evidence of efficacy in two mouse models of the disease, as well as preliminary data on the applicability of their approach to cell therapy in nonhuman primates.(3)
A Superior Protocol
The authors had previously reported(4) a protocol for deriving mesencephalic DA neurons by nudging them through an intermediary stage as midbrain floor plate precursor cells -- the developmental topos through which cells are thought to acquire neural progenitor characteristics. This protocol had subsequently been independently validated;(5) it involves inhibition of SMAD signalling by inhibiting BMP4 using Noggin (an inhibitor of BMP4) and activating the Lefty/Activin/TGFβ pathway with the drug SB431542. Two lines each of human ESC and iPS cells were treated using a modified version of this strategy to derive DA neurons. This approach proved superior to the currently-standard protocol(6) of moving such cells through a neural rosette intermediate for DA neuron derivation, generating a higher percentage of tyrosine hydroxylase-expressing neurons, expression of markers of the developing mesencephalon and (importantly) much lower generation of serotonergic neurons.(3)
Dopaminergic Phenotype
One weakness common to many studies of derivation of differentiated cells from pluripotent precursors has been the almost exclusive reliance on cell-surface markers as indicators of ultimate fate, casting uncertainty over our understanding of such cells and the interpretation of both therapeutic benefits and adverse reactions from the use of such cells in models of injury and disease. The authors loaned substantial credibility to their results by showing that their human ESC- and iPS-derived DA cells recapitulate multiple phenotypic characteristics of native substantia nigra DA neurons and nigral neurons grown from early postnatal mice, including not only high expression of their mature neuronal markers, but their specific electrophysiological phenotype and extensive fiber outgrowth.(3)
Success and Safety In Vivo
The investigators next tested cells derived either from their own modified dual SMAD-inhibition protocol or the rosette method in vivo. Based on previous work, the investigators performed all transplantation during cell cycle exit; such cells proved to survive effectively in intact rodents, and were next tested by unilateral striatal injection into the 6-hydroxy-dopamine (6-OHDA) lesion model of PD. Because it has proven challenging to develop differentiated cells that exhibit robust growth and strong engraftment in local neuronal tissue without leading to "off-target" differentiation into undesired cell types, or tumorigenic overgrowth, the radically immunodeficient NOD-SCID IL2Rgc null mouse was used for preclinical safety and efficacy studies; this strain "efficiently supports xenograft survival with particular sensitivity for exposing rare tumorigenic cells."(3) Moreover, neuronal cultures were grafted "as is" rather than following additional cell purification, to give maximum opportunity for any insufficiently- or improperly-differentiated cells to cause overgrowth.
At four and a half months post-transplant, the fates of cells derived from the two protocols in vivo were dramatically different. In animals engrafted with DA neurons derived using the standard rosette method, administration of amphetamine led to circling motion behavior, caused by the drug's stimulation of the imbalanced populations of DA neurons surviving on the two sides of the brain following unilateral 6-OHDA lesioning. In the other group, engraftment with cells derived using the floor plate method rescued this behavior. In animals treated with rosette-derived DA neurons, the graft site exhibited few human-derived dopaminergic neurons, was riddled with proliferating Ki-671+
cells, and exhibited "massive neuroal overgrowth";(3) the equivalent site in animals receiving floor plate-derived cells was well-populated with human-derived DA neurons, and the site's borders were clearly-defined, <1% of total cells actitively proliferating.(3)
To eliminate artifacts of reliance on the immunodeficient mouse model, these experiments were repeated in rats immunosuppressed using cyclosporin A, this time comparing neural grafts using cells derived wth their new protocol to sham treatment. Again, high numbers of transplanted DA neurons engrafted, survived, and branched out to integrate with surviving tissue without overgrowth; gene expression profiles suggested the presence of both nigral and ventral tegmental area DA neurons. Treatment again eliminated rotational behavior following amphetamine administration, and also alleviated deficits in two domains that manifest without drug-induced overactivation of surviving DA neurons in lesioned mice: forelimb akinesia on the stepping test, and the specific use of the forelimb using the cylinder test (see Figure 1).(3) In this model too, <1% of total cells had improperly differentiated into serotonergic neurons, and the few GFAP1 glial cells were host-derived.(3)
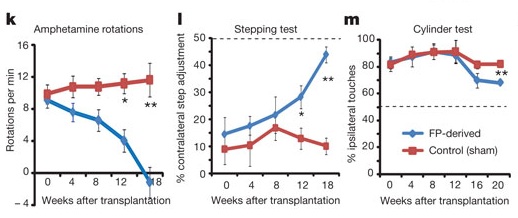
Figure 1. Floor-Plate (FP)-Derived Dopaminergic Neuron Grafts from Human ESC Rescue Motion Disorders in 6-OHDA-Lesioned Rats. Reproduced from (3).
Preliminary Promise in Primates
Finally, the investigators performed a minimal test the viability of their approach in a nonhuman primate model, genetically closer to our own species and better reflecting the much larger number of neurons that would be required for human therapeutic purposes relative to the much smaller rodent models. Their system allowed for ready derivation of 5 x 107 DA neurons for testing, and following MPTP lesioning of the substantia nigra, each of two adult Rhesus monkeys received a total of six transplants of 1.25 x 106 cells in each of three sites spanning the posterior caudate and pre-commissural putamen on each side of the brain. One month later, GFP expression revealed high numbers of surviving mesencephalic DA neurons coexpressing the human-specific cytoplasmic marker SC-121 at each site, with fibers branching out of the graft cores for up to 3mm into surrounding host brain tissue. The sole note of caution was the suggestion of incomplete immunosuppression, based on the presence of Iba1+ host microglial cells in the grafts.(3)
Continuing, Perfecting, and Integrating a Rejuvenation Biotechnology
As the researchers conclude, their
novel FP-based [pluripotent stem cell] differentiation protocol faithfully recapitulates midbrain DA neuron development. ... Importantly, our study establishes a means of obtaining a scalable source of FOXA21/ TH1 neurons for neural transplantation—a major step on the road towards considering a cell based therapy for Parkinson’s disease. Excellent DA neuron survival, function and lack of neural overgrowth in the three animal models indicate promise for the development of cell-based therapies in Parkinson's disease.(3)
The most immediate next step in development of this potential therapy is to test the ability of these grafts to rescue the motion disorders of MPTP-lesioned nonhuman primates. Were such cells to prove themselves in this model, clinical trials in humans suffering with PD from aging-induced DA neuronal losses would be fully justified, based on the limited positive results already achieved in cell therapy for this disorder: engraftment with pure populations of pristine DA neurons should deliver the benefits previously observed using mixed fetal/embryonic tissue, without the dyskinesic side-effects apparently flowing from contamination with serotonergic cells.(2)
Once these cells have proven their potential in humans, yet further refinement of the protocol can be expected to more fully alleviate symptoms. In this study,(3) as in the previous human(0) and preclinical work, transplanted cells have been injected directly into the striatum, where the loss of DA is acute. To fully restore the intact circuitry of the youthful, fully-functioning dopaminergic system will require protocols for the orthotopic transplantation of such cells into the neurodegenerative substantia nigra, recapitulating the physiological dopaminergic innervation of the striatum from this graft core.
Additionally, in the earlier trials, transplanted cells developed intraneuronal aggregates composed of α-synuclein, characteristic of the aging and particularly the PD brain, at a rate that appears to be relatively slow but perhaps faster than that in native, undiseased neurons;(7) indeed, there is strong evidence that accumulation of such aggregates play a key role in PD, and their temporal neuropathological staging is initiated in the aging hindbrain, remote from the substantia nigra, and gradually spreads forward toward it. This more widespread neuropathology is thought to be a major contributor to the many nondopaminergic, levodopa-refractory symptoms of PD.(8) Brain rejuvenation will require clearance of these aggregates to maintain the benefits of transplanted cells and to eliminate these troublesome and disabling symptoms, such as through the use of novel xenohydrolases.(9)
A key barrier to the use of human pluripotent stem cells to rejuvenate the aging and neurodegenerative brain appears to have been broken; it is now our task to press on, treating Parkinson's disease and ultimately ending the age-related degeneration of the human brain.
References
0: Lindvall O, Björklund A. Cell therapy in Parkinson's disease. NeuroRx. 2004 Oct;1(4):382-93. Review. PubMed PMID: 15717042; PubMed Central PMCID: PMC534947.
1: English K, Wood KJ. Immunogenicity of embryonic stem cell-derived progenitors after transplantation. Curr Opin Organ Transplant. 2010 Dec 9. [Epub ahead of print] PubMed PMID: 21150615.
2: Politis M, Wu K, Loane C, Quinn NP, Brooks DJ, Rehncrona S, Bjorklund A, Lindvall O, Piccini P. Serotonergic neurons mediate dyskinesia side effects in Parkinson's patients with neural transplants. Sci Transl Med. 2010 Jun 30;2(38):38ra46. PubMed PMID: 20592420.
3: Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011 Nov 6. doi: 10.1038/nature10648. [Epub ahead of print] PubMed PMID: 22056989.
4: Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009 Mar;27(3):275-80. Epub 2009 Mar 1. Erratum in: Nat Biotechnol. 2009 May;27(5):485. PubMed PMID: 19252484; PubMed Central PMCID: PMC2756723.
5: Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, Schapira AH, Gwinn K, Hardy J, Lewis PA, Kunath T. Parkinson's disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun. 2011 Aug 23;2:440. doi: 10.1038/ncomms1453. PubMed PMID: 21863007.
6: Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008 Jan 15;22(2):152-65. Erratum in: Genes Dev. 2008 May 1;22(9):1257. PubMed PMID: 18198334; PubMed Central PMCID: PMC2192751.
7: Braak H, Del Tredici K. Assessing fetal nerve cell grafts in Parkinson's disease. Nat Med. 2008 May;14(5):483-5. PubMed PMID: 18463652.
8: Lang AE, Obeso JA. Challenges in Parkinson's disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol. 2004 May;3(5):309-16. Review. PubMed PMID: 15099546.
9 : Mathieu JM, Schloendorn J, Rittmann BE, Alvarez PJ. Medical bioremediation of age-related diseases. Microb Cell Fact. 2009 Apr 9;8:21. PubMed PMID: 19358742; PubMed Central PMCID: PMC2674406.
View the full article
ImmInst
03 Dec 2011
To develop an unbreachable defense against cancer, SENS Foundation is pursuing the WILT (Wholebody Interdiction of Lengthening of Telomeres) strategy (otherwise OncoSENS) of systematically deleting genes essential to the cellular telomere-maintenance mechanisms (TMM) from all somatic cells, while ensuring ongoing tissue repair and maintenance through periodic re-seeding of somatic stem-cell pools with autologous TMM-deficient cells whose telomeres have been lengthened ex vivo. In addition to the deletion of one or more genes coding for essential element(s) of the telomerase holoenzyme, success will also require the deletion of some essential element of the machinery [responsible] for the Alternative Lengthening of Telomeres (ALT) phenomenon, observed in a minority of cancer cells.
One of the characteristic phenotypic features of cells utilizing the ALT mechanism is the presence of telomeric DNA and the telomere binding proteins TERF1 and TERF2 in close association with a subset of the cell's promyelocytic leukaemia protein (PML) nuclear bodies. PML nuclear bodies are spherical nuclear structures which, in normal cells, are involved in DNA repair, senescence, apoptosis, and other functions. But their association with telomeric chromatin is rare, except in ALT cells, where they are instead hallmarks of the phenomenon; this has led to such assemblies being designated "ALT-associated PML bodies" (APBs). APBs not only contain recombination proteins demonstrated to be essential to telomere maintenance in ALT cells, but appear themselves to be involved in telomere recombination(1) and to coincide spatially and temporally with telomeric DNA synthesis.(2,3) Therefore, some have hypothesized that APBs may be the nexus of the “Alternative Lengthening of Telomeres” in such cells.
To test whether APBs might indeed be causally involved in the TMM of ALT cells, a team of German researchers devised the novel approach of generating APBs artificially, by recruiting APB subcomponents to telomeric or pericentric DNA, and observing the effects on assembly of APB-like structures and on telomere elongation.(4) To do this, the investigators generated fusion proteins of several APB constituents with the bacterial LacI repressor protein and fluorescent binding proteins as markers, and then exploited LacI's high-affinity binding with the operator region of lac operons to tether these proteins to the repeats of lac operator sequences that are stably integrated into the genome of U2OS osteosarcoma cell lines. To reveal the effects of chromosomal localization on such proteins' ability to form APBs and on the abilities of such experimentlaly-induced entities, the researchers tested the effects of APB constituent protein tethering in two different U2OS cell lines: one (F6B2) with such operator sequences integrated adjacent to telomeric DNA, and another (F42B8) whose sequences were localized pericentrically. Thus, transfection of one or the other cell line with LacI-APB component fusion constructs resulted in the tethering of the various APB component proteins at locations proximate to, or remote from, telomeric DNA.(4)
Bring a Friend
When tethered to telomeric DNA, LacI fusion constructs containing either of the two proteins (PML protein or Sp100) present in PML nuclear bodies, induced recruited the complementary protein in turn; tethering of "empty" fluorescent-labeled LacI constructs yielded no such results.(4) The bringing-together of these proteins at telomeric DNA induced the formation of apparent APBs, as suggested by the observations that (a) the PML protein assumed the same cap-like structures surrounding telomere repeats observed in APBs; (b) the structures appeared almost entirely localized with single telomere structures; and © the location of the recruited endogenous proteins appeared enriched with several SUMO isoforms, consistent with the presence of SUMO modifications of these proteins in endogenous PML nuclear bodies.(5)
Sussing Out SUMO
Indeed, modification of at least some APB constituents by SUMO proteins is known to be essential to the formation of APBs: PML protein contains a SUMO-interacting motif (SIM) that is not involved in SUMOylation but that that is required for PML nuclear body formation.(5) Again mirroring observations in native APBs, telomeric recruitment of LacI constructs with several SUMO isoforms led to colocalization of native PML, Sp100, and Rad17 (all present in endogenous APBs), again consistent with APB formation. A series of tests using constructs containing SUMO1 mutants either incapable of the noncovalent interactions with SIMs observed in the APBs in endogenous ALT cells, or incapable of covalent attachments, demonstrated that the role of this SUMO protein in the formation of the de novo APBs involves the former kind of interactions,(4) consistent with the need for noncovalent SIM binding in the formation of native PML nuclear bodies.
On the other hand, several telomere-associated proteins present in APBs in ALT (including TRF1, TRF2, and Rap1) do require SUMOylation, by the SUMO E3 ligase MMS21, and here again the investigators were able to use their tethering system to recapitulate observations of native APBs. Tethering of MMS21 to telomereic or pericentric DNA was "highly efficient in promoting APB assembly," recruiting substantial increases in colocalized PML protein. But importantly, only those MMS21-tethered telomeres with colocalized PML also colocalized with the APB component Rad9. This was hightly similar to what they observed in native APBs: "The vast majority (98%) of endogenous APBs (defined as colocalization of PML and TRF2) contained Rad9, and only 0.9% of the telomeres marked by TRF2 had Rad9 but no PML."(4) This is especially noteworthy, since there is evidence(5) that MMS21 plays a role in DNA repair, and the ALT TMM itself exploits (telomeric) DNA repair.
Back of the Line, Please
Inducing TRF1, TRF2, Rad9, NBS1, and MMS21 recruitment to telomeric DNA also increased formation of APBs.(4) On the other hand, tethering of two other APB constituents (Rad51 or Rad17) to telomeric DNA led to little (Rad51) or no (Rad17) recruitment of PML.(4) Despite this, endogenous Rad17 -- like NBS1 and Rad9 -- was enriched in the de novo APBs formed following PML tethering, consistent with native APBs. The authors note that a previous report (5) had found that knockdown or Rad51 with siRNA in U2OS cells had no effect on APB formation; combined, these results suggest that these repair factors may only be recruited to APBs after the initial assembly of other constituent proteins.
The Road to Perdition -- and a Bridge to Nowhere
In addition to observations at telomeric DNA, APB component proteins were also recruited when the researchers tethered PML to the pericentric lac operator sequences in the F442B8 U2OS line; in fact, these components were recruited "to a similar or even higher degree than at the telomeric sites." Again in general agreement with observations using telomerically-tethered proteins, recruiting MMS21 to pericentric DNA also led to elevated colocalization of APB proteins, whereas tethering of Rad51 at this locus did not.(4)
But the purpose of the investigation was not to determine if APBs could induced to form at these loci, but to determine the function of these structures: whether APBs play a causal role in telomere maintenance in ALT cells, or are merely phenotypic hallmarks of the ALT phenomenon. The investigators therefore tested the ability of de novo APBs to lengthen telomeres in a manner consistent with the ALT TMM.
When PML was tethered to telomeric chromosomal loci, the resulting de novo APBs were associated with elevated phosphorylated γ-H2AX histone. Phosphorylated γ-H2AX is a component of APBs, and indicator of double-strand break (DSB) repair, which is involved in the ALT TMM.(4) Similarly, the generation of de novo APBs at telomeric chromosomes was associated with increased non-replicative DNA synthesis, as detected by elevated pulsed 5-bromo-2′-deoxyuridine (BrdU) incorporation at the limited number of foci where it occurs during non-replicative phases of the cell cycle. Such increases in DNA synthesis, arising in association with the formation of APBs at telomeric DNA, are consistent with telomere lengthening. Again, a fluorescence in situ hybridization (FISH) probe was used to detect the percentage chromosomal ends too short to register on the probe, and thus primed for telomere extension by the ALT TMM. Tethering PML to telomeric DNA reduced the fraction of chromosomes with indetectably-short telomeres from ~43% to ~19%, suggesting the extension of critically-short telomeres; by contrast, tethering "empty" fluorescent-labeled LacI constructs to telomeric DNA led to no such increase.(4)
And importantly, none of these phenomena -- neither increased phosphorylated γ-H2AX, nor increased non-replicative BrdU incorporation, nor a reduction in indetectably-short telomeres -- appeared subsequent to APB formation at pericentric lac operator sequences. Thus, these phenomena -- each consistent with the lengthening of telomeres by an ALT-like mechanism -- were specific to the presence of APBs at telomeric DNA.(4)
Pause del Silenzio
It is worth here quoting the authors' own conclusions at length:
[Our findings] establish APBs as functional intermediates of the ALT pathway [my emphasis]. ... Accordingly, we conclude that the formation of bona fide APBs promotes the extension of the telomere repeat sequence by a DNA- repair-coupled synthesis process. Our study does not provide information on the nature of the telomere repeat template used for synthesis ... Furthermore, given the large number of partially contradicting results in the literature, it is well conceivable that different telomerase-independent mechanisms for telomere repeat extension exist. ... Thus, it will be important to further dissect the exact combination of protein factors that are sufficient to trigger telomere extension in APBs and to investigate the effects of their presence or absence. We anticipate that the experimental approach introduced here ... will allow us to precisely identify all protein components that are sufficient to form a telomeric PML-NB subcompartment structure, as well as the additional factors needed to induce telomere extension at these sites. This will serve to select protein targets for inhibiting telomere extension, and thus cell proliferation, in tumors that make use of the ALT pathway.(4)
All of the known constituents of APBs play a role in normal cellular metabolism. But full implementation of the WILT strategy will require not only that we "select [APB] protein targets for inhibiting telomere extension," but that one or more element of the ALT TMM be irreversibly disabled, in all of the cells of the body (and as a major intermediate goal, in those tissues in which ALT cancers most commonly arise). Thus, success with WILT will require a detailed elaboration of all the structures responsible for the lengthening of telomeres in ALT cells -- those discovered and undiscovered, constitutive of APBs and not -- and an understanding of their role in telomere lengthening in disease, as well as physiological functions in health. By providing significant support for the role of APBs in telomere extension, the beginnings of further understanding of the process of APB assembly and the roles of some constituent proteins in the APB TMM, and new tools for dissecting APB structure and function, Chung et al have taken us a significant step toward this benchmark, and thus toward a decisive cure for malignant disease.
References
1: Draskovic I, Arnoult N, Steiner V, Bacchetti S, Lomonte P, Londoño-Vallejo A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc Natl Acad Sci U S A. 2009 Sep 15;106(37):15726-31. Epub 2009 Aug 26. PubMed PMID: 19717459; PubMed Central PMCID: PMC2747187.
2: Nabetani A, Yokoyama O, Ishikawa F. Localization of hRad9, hHus1, hRad1, and hRad17 and caffeine-sensitive DNA replication at the alternative lengthening of telomeres-associated promyelocytic leukemia body. J Biol Chem. 2004 Jun 11;279(24):25849-57. Epub 2004 Apr 9. PubMed PMID: 15075340.
3:Wu G, Lee WH, Chen PL. NBS1 and TRF1 colocalize at promyelocytic leukemia bodies during late S/G2 phases in immortalized telomerase-negative cells. Implication of NBS1 in alternative lengthening of telomeres. J Biol Chem. 2000 Sep 29;275(39):30618-22. PubMed PMID: 10913111.
4: Chung I, Leonhardt H, Rippe K. De novo assembly of a PML nuclear subcompartment occurs through multiple pathways and induces telomere elongation. J Cell Sci. 2011 Nov 1;124(Pt 21):3603-18. Epub 2011 Nov 1. PubMed PMID: 22045732.
5: Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. The mechanisms of PML-nuclear body formation. Mol Cell. 2006 Nov 3;24(3):331-9. PubMed PMID: 17081985; PubMed Central PMCID: PMC1978182
6: Potts PR, Yu H. Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol Cell Biol. 2005 Aug;25(16):7021-32. PubMed PMID: 16055714; PubMed Central PMCID: PMC1190242.
View the full article
ImmInst
24 Dec 2011
| ||||||

View the full article
ImmInst
02 Jan 2012
Immunotherapy targeting the age-related accumulation of extracellular aggregates, in the form of ß-amyloid, is the first rejuvenation biotechnology to reach Phase III human clinical trials. The promise of this therapy for the treatment and prevention of Alzheimer's disease (and ultimately, of so-called "normal" brain aging) has sparked an interest in utilizing the same approach for other forms of aging damage, including the clearance of aggregated intracellular proteinaceous aging damage. Notably, as we have reviewed in a series of four previous posts, recent years have seen the appearance of a rising number preclinical studies of therapeutic vaccines targeting pathological tau species accumulating in the brains and spinal cords of transgenic rodent models of tauopathic neurodegeneration. These studies have reported -- somewhat surprisingly -- the antibody-mediated clearance of these primarily intracellular aging lesions, accompanied by functional improvements in treated animals.(4-8) These two forms of structural damage are major contributors to the age-related degeneration of the brain, whether it leads to frank dementia or to the diagnostic euphemism of "normal" age-related cognitive decline, and novel therapeutics to effect the removal of both from aging neurons will be key elements of a comprehensive panel of rejuvenation biotechnologies.
One limitation to the existing studies of vaccines targeting pathological tau has been that they have tended to initiate therapy before, at, or only very shortly after the appearance of tau pathology in the murine rodent system, and of the onset of cognitive or motor deficits -- a luxury which we cannot expect to be afforded in treating cases of frank dementia related to these inclusions, and that may not reflect the condition of the brain even prior to the appearance of gross cognitive and functional deficits in aging humans. Further complicating these matters, the combination of early intervention and the very rapid progression of tau pathology in these models has meant that these studies have given only limited information on the effects of the onset or later clearance of neurofibrillary tangles (NFT), which are the most prominent and mature form of tau pathology to appear in the brain during aging, in Alzheimer's disease, and with the genetic tauopathies. A group headed by Dr. Lars Ittner of the Laboratory for Translational Neurodegeneration at the University of Sydney, Camperdown has now provided the first evidence on these unresolved questions, furthering the case for human therapies targeting pathological tau.
Ittner's group performed their studies in pR5 mice, which express the mutant human tau species P301L, which is responsible for the human dementing tauopathy frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17). These mice begin to accumulate tau that has been hyperphosphorylated at different epitopes early in life, but phosphorylation of the NFT-associated Ser422 and Ser396/S404 (the paired helical fragment-1 (PHF-1) epitope) first begins to appear at the 6 mo mark, accompanied by NFT pathology, which progresses thereafter throughout their shortened lifespans. The investigators accordingly engineered a vaccine targeting these NFT-associated species by linking a human tau peptide comprising phosphorylated PHF-1 Ser396/Ser404 to the immunogenic carrier protein KLH. This vaccine was then administered to animals before (4 mo), shortly after (8 mo), and long after (18 mo) the 6-month age of NFT onset, accompanied by complete Freund's adjuvant, and later followed by further adjuvant booster.(1)
By 17 mo, hippocampal and amygdalic neurons of untreated mice exhibited high levels of antibody staining for phorphoylated NFT-associated tau epitopes; even more notably, staingin was also high in the dystrophic neurites in subregions of these areas. Immunization targeting NFT tau epitopes had no effect on expression or distribution of either human mutant or total (transgenic plus native murine native) tau, but did result in robust (≥1:1000) antigen titers within four months of vaccination. Notably, even untreated pR5 mice exhibited low levels of such titers, while no such antibodies were evident in isochronic wild-type mice; this suggests a (weak) intrinsic immune response against the presence of aberrant tau species in mice bearing them.(1) The existence of such a response is consistent with previous findings in aging humans,(3) Alzheimer's disease patients,(3) and transgenic murine tauopathy.(2)
Tau-targeting vaccination substantially reduced the subsequent burden of pathological tau in neurons and dystrophic neurites, no matter when treatment was initiated:
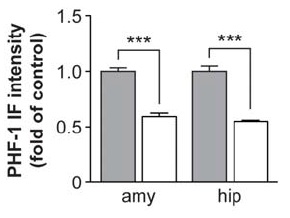
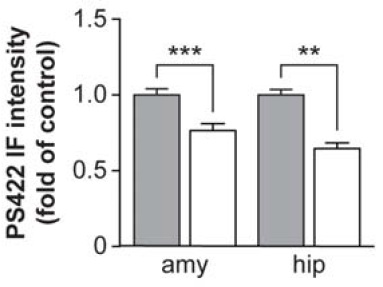
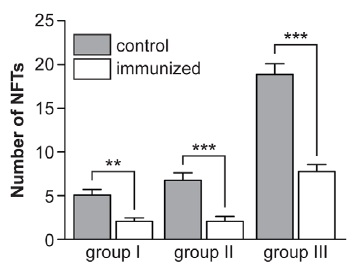
Figure 1: Vaccination targeting pathological tau species reduces the further spread of antibody-labeled PHF-1 (left) and tau phosphorylated at Ser422 (center) when initiated shortly after NFT appearance; when administered before (Group I), shortly after (Group II), or long after (Group III) their appearance, it similarly impeded the appearance of Gallyas silver-stained NFTs per se (right),. Reproduced from (1) under the terms of Creative Commons License.
Limitations, and their Supercedance
One key limitation to this study is that no cognitive or motor function data are presented. In prior studies, however, preventive and therapeutic vaccination in similar murine models of tau neuropathy has led to relative improvements in such functions, proportional to the reduction in neuropathology.
Another limitation is of interpretation of the results, and possibly of the actual effects of the vaccine therapy itself. The authors note that the burden of NFT in treated animals from Group III -- aged 22 mo at autopsy, but whose immunization was delayed until 18 mo of age (ie, a full year after the onset of neuropathology) -- was similar to that in untreated mice shortly before (Group I) or shortly after (Group II) that onset at autopsy. Because autopsied Group II animals were of similar age to the the age at which vaccination was initiated in Group III, and unvaccinated Group II animals' neuropathology burden was similar to that present months after treatment in animals that were initially treated at a similar age, the authors suggest that the apparently similar NFT burdens after several months of vaccination in the latter group suggest that the treatment had arrested the further accumulation of tau pathology following vaccination, rather than having actively cleared it. However, as they also note, it is also possible that their tau vaccine did remove existing tau inclusions, but that the rate of active clearance of tau pathology was equal to the rate of new NFT accumulation.(1) This is quite plausible, granted the very rapid progression of tau pathology in these animals when left untreated, and even more so granted their relatively brief period of treatment: Group III was sufficiently far into their shortened life expectancy as to necessitate abbreviated (6 mo) therapy, relative to the 9-10 mo afforded to Groups I and II. Thus, a longer period of antibody-mediated clearance might have afforded a more thorough therapeutic effect.
At a minimum, even a vaccine that only arrested the burden of tau pathology could contribute to the prevention of significant age-related cognitive decline if administered in middle age. And in aging humans, the development of neurofibrillary tangles occurs over the course of decades, rather than months as in these animals; thus, even a moderate rate of true clearance could in principle be sufficient to lead to a net reduction in NFT burden in humans, and thus to the reversal of one component of brain aging.
In either case, several strategies could be explored to enhance the efficacy of such a vaccine. The most obvious is the administration of a series of booster shots, to increase therapeutic antibody titers or to restore putatively flagging numbers in the months (or in humans, perhaps years) after initial vaccination. Note that the investigators only collected a single data point on antibodies targeting pathological tau, taken relatively soon (4 mo) after a single vaccination at a single antigen dose, following which the same, single round of vaccination was left to work for a further 6 (Groups 1 and II) or 2 (Group III) months in younger and older animals, respectively; this clearly leaves open the potential that a series of rounds of treatment could have been more effective.
Another potential way to enhance the efficacy of a similar vaccine in humans would be to fortify the lysosome with novel hydrolytic enzymes, to enhance its capacity to clear out aggregates which might be targeted to the lysosome by anti-phospho-tau antibodies. While the involvement of lysosomal degradation of aberrant tau species in the therapeutic effects of immunotherapy was not evaluated in this study,(1) such an effect is consistent with the clearance of intraneuronal phospho-tau in these vaccination studies, and with much other data. Vaccination of a Parkinson's disease transgenic mouse model with α-synuclein, similarly, leads to a reduction of intracellular α-synuclein aggregates;(11) several studies have found that immunotherapy targeting β-amyloid leads to a reduction in intraneuronal forms of the aggregate, and work in Gunnar Gouras' laboratory, reviewed in an earlier post, finds that antibodies against Aβ can be internalized in AD neuronal culture models of Aβ accumulation and clear intraneuronal Aβ aggregates via the endosomal–lysosomal pathway.Additionally, the authors of the current study(1) also note that immunized K257T/P301S tauopathy mice exhibit changes in cathepsin levels, consistent with lysosomal degradation of pathological tau;(6) in this context, it is notable that the lysosomal-enhancing drug PADK (see a previous post) removes PHF-1 from hippocampal slices, in association with a normalization of impaired synaptic integrity.(10,11) I will note that the authors of the current study(1) do argue against such a mechanism, on the basis that "total tau levels remained unchanged upon immunization of JNPL3 mice [(5) below], and staining with total tau antibodies was comparable in immunized and control pR5 mice (this study), arguing against overtly increased degradation of tau" -- but of course, this does not argue against specific degradation of pathological tau species targeted by immunotherapy in their study.(1)
An additional possibility is that the clearance of other intraneuronal aggregates from aging neurons may itself facilitate the additional proteolytic or lysosomal removal of phospho-tau species. The relationship between the accumulation and neurotoxicity of Aβ and aberrant tau is complex,(13) but reducing one will almost certainly reduce the deleterious effects of the other; and, in particular, immunization against Aβ has been reported to lead to the clearance of early phospho-tau species.(14 (and see (15)) This adds a further argument in favor of a comprehensive approach to brain aging and Alzheimer's disease, suggesting synergistic rather than merely additive effects of combining therapies targeting Aβ in addition to malformed tau.
Finally, it is also possible that future vaccines might be more specifically targeted to especially pathologically important tau species, or be designed to elicit antibodies with higher avidity for their target. All of these avenues deserve exploration in future studies of this promising therapeutic approach.
Breaking from a March to a Run
This latest report(1) adds to a series of previous successful preclinical studies(4-8) of immunotherapies targeting pathological tau species for therapeutic clearance. This new study is notable for marking the entry of a new group of investigators into the tau vaccine space, suggesting that this area of rejuvenation biotechnology is attracting an increasing number of independent research teams. While limited in scope and in design, the new study adds to prior knowledge not only by testing a new vaccine, but importantly by evaluating its effects in animals in whom damage from tau inclusions was already extensive. While cognitive and motor functions were not evaluated, the clear ability of this vaccine to arrest -- and perhaps to reverse -- the burden of a life-long, accelerated deposition of aggressive pathological tau species offers promise for human translation into a treatment for genetic tauopathies, Alzheimer's disease, and the "normal" age-related decay of cogntitive, emotional, and neurological function that is only now finally being recognized as a disorder meriting therapy -- and ultimately, cure. The gaps in this study are opportunities for further experimentation, further refinement, further learning, and further therapeutic development; it marks progress, bringing us closer to the goal of a comprehensive panel of rejuvenation biotechnologies and the rescue of aging bodies and minds.
References
1: Bi M, Ittner A, Ke YD, Götz J, Ittner LM. Tau-Targeted Immunization Impedes Progression of Neurofibrillary Histopathology in Aged P301L Tau Transgenic Mice. PLoS One. 2011;6(12):e26860. Epub 2011 Dec 8. PubMed PMID: 22174735; PubMed Central PMCID: PMC3234245.
2: Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006 Oct;63(10):1459-67. PubMed PMID: 17030663.
3: Rosenmann H, Meiner Z, Geylis V, Abramsky O, Steinitz M. Detection of circulating antibodies against tau protein in its unphosphorylated and in its neurofibrillary tangles-related phosphorylated state in Alzheimer's disease and healthy subjects. Neurosci Lett. 2006 Dec 20;410(2):90-3. PubMed PMID: 17095156.
4: Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006 Oct;63(10):1459-67. PubMed PMID: 17030663.
5: Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007 Aug 22;27(34):9115-29. PMID: 17715348 [PubMed - in process]
6: Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010 Aug;224(2):472-85. Epub 2010 May 28. PubMed PMID: 20546729.
7: Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem. 2011 Aug;118(4):658-67. doi: 10.1111/j.1471-4159.2011.07337.x. Epub 2011 Jul 1. PubMed PMID: 21644996.
8: Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010 Dec 8;30(49):16559-66. PubMed PMID: 21147995.
9: Wisniewski T, Sigurdsson EM. Murine models of Alzheimer's disease and their use in developing immunotherapies. Biochim Biophys Acta. 2010 Oct;1802(10):847-59. Epub 2010 May 13. PubMed PMID: 20471477; PubMed Central PMCID: PMC2930136.
10: Butler D, Brown QB, Chin DJ, Batey L, Karim S, Mutneja MS, Karanian DA, Bahr BA. Cellular responses to protein accumulation involve autophagy and lysosomal enzyme activation. Rejuvenation Res. 2005 Winter;8(4):227-37. PubMed PMID: 16313222.
11: Bendiske J, Bahr BA. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis--an approach for slowing Alzheimer disease? J Neuropathol Exp Neurol. 2003 May;62(5):451-63. PubMed PMID: 12769185.
12: Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005 Jun 16;46(6):857-68. PMID: 15953415 [PubMed - indexed for MEDLINE]
13: Ittner LM, Götz J. Amyloid-β and tau--a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011 Feb;12(2):65-72. Epub 2010 Dec 31. Review. PubMed PMID: 21193853.
14: Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004 Aug 5;43(3):321-32. PMID: 15294141 [PubMed - indexed for MEDLINE]
15: Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006 Jan 20;281(3):1599-604. Epub 2005 Nov 10. PMID: 16282321 [PubMed - indexed for MEDLINE]
View the full article
 Avatar of Horus
18 Sep 2015
Avatar of Horus
18 Sep 2015
In late 2008, we reviewed then-unpublished work by universal constituent of the abnormal tissue deposits in amyloidosis, including Alzheimer disease". (2) As we reviewed:
The promise
A quarter century ago, Pepys suggested that because circulating SAP is believed to exist in a state of dynamic equilibrium with the SAP in amyloid deposits, lowering circulating SAP might lead plaque SAP to dissociate, leading to the breakup of the integrity of the plaque and ultimate clearance of amyloid deposits.
Early in this century, Pepys' team began ... search[ing for] a small molecule that might inhibit the binding of SAP in Abeta, and came across one that was particularly effective: R-1-[6-[R-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl] pyrrolidine-2-carboxylic acid, or CPHPC, which "also crosslinks and dimerizes SAP molecules",([3]) blocking the binding face of the molecule in the process. They quickly moved from this in vitro finding through animal studies for efficacy and toxicity, and the initial results being favorable, moved into pilot human studies.([4])
In 2002, Pepys reported that adminstration of IV CPHPC over the course of 2 days into 8 amyloidosis patients resulted in almost total removal of SAP from the circulation, apparently through "very rapid" hepatic clearance, since tracer studies found a large amount of SAP in the liver of one patients 6 h after initiating treatment.([4]) They quickly sent CPHC sailing down the drug-development pipeline, to results that, although still preliminary, were extremely exciting.
We reviewed some preliminary findings from subsequent pilot studies in light chain (AL) amyloidosis and other diseases, some of which were later reported and elaborated at SENS4 -- including very brief allusion to some work on AD patients.
...
Some developments on this approach were reported recently, not just CPHPC, but also targeting SAP directly with antibody:
...
And, about the Apcs protein, also known as the SAP, serum amyloid P component, which seems to play a role in the various forms of amyloidosis diseases:
...
And, last but not least, a recent development, about an approach using the SAP protein for the removal of amyloid deposits:Improving treatment for systemic amyloidosis
July 16, 2015
http://medicalxpress...myloidosis.html
A potential new approach to treat systemic amyloidosis, invented at UCL and being developed by GlaxoSmithKline (GSK), marks the start of a successful and innovative academic-industry collaboration.
The first in human clinical trial of a novel investigational drug intervention for patients with systemic amyloidosis has established proof of mechanism. Results in the first 15 patients treated with a therapeutic partnership of a small chemical molecule and a large biological molecule (an antibody) are published in the New England Journal of Medicine. Further clinical testing is in progress and a phase II trial to explore efficacy and safety is planned.
...Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component
Richards DB1, Cookson LM, Berges AC, Barton SV, Lane T, Ritter JM, Fontana M, Moon JC, Pinzani M, Gillmore JD, Hawkins PN, Pepys MB.
N Engl J Med. 2015 Sep 17;373(12):1106-14. doi: 10.1056/NEJMoa1504942. Epub 2015 Jul 15.
http://www.ncbi.nlm....pubmed/26176329
Abstract
BACKGROUND: The amyloid fibril deposits that cause systemic amyloidosis always contain the nonfibrillar normal plasma protein, serum amyloid P component (SAP). The drug ®-1-[6-[®-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid (CPHPC) efficiently depletes SAP from the plasma but leaves some SAP in amyloid deposits that can be specifically targeted by therapeutic IgG anti-SAP antibodies. In murine amyloid A type amyloidosis, the binding of these antibodies to the residual SAP in amyloid deposits activates complement and triggers the rapid clearance of amyloid by macrophage-derived multinucleated giant cells.
METHODS: We conducted an open-label, single-dose-escalation, phase 1 trial involving 15 patients with systemic amyloidosis. After first using CPHPC to deplete circulating SAP, we infused a fully humanized monoclonal IgG1 anti-SAP antibody. Patients with clinical evidence of cardiac involvement were not included for safety reasons. Organ function, inflammatory markers, and amyloid load were monitored.
RESULTS: There were no serious adverse events. Infusion reactions occurred in some of the initial recipients of larger doses of antibody; reactions were reduced by slowing the infusion rate for later patients. At 6 weeks, patients who had received a sufficient dose of antibody in relation to their amyloid load had decreased liver stiffness, as measured with the use of transient elastography. These patients also had improvements in liver function in association with a substantial reduction in hepatic amyloid load, as shown by means of SAP scintigraphy and measurement of extracellular volume by magnetic resonance imaging. A reduction in kidney amyloid load and shrinkage of an amyloid-laden lymph node were also observed.
CONCLUSIONS: Treatment with CPHPC followed by an anti-SAP antibody safely triggered clearance of amyloid deposits from the liver and some other tissues. (Funded by GlaxoSmithKline; ClinicalTrials.gov number, NCT01777243.).
http://clinicaltrial...how/NCT01777243


